


Aberrant activation of autoimmunity may influence the bad impact on the liver. The representative entities of autoimmune liver disease include autoimmune hepatitis (AIH), primary biliary cholangitis (PBC), and primary sclerosing cholangitis (PSC).
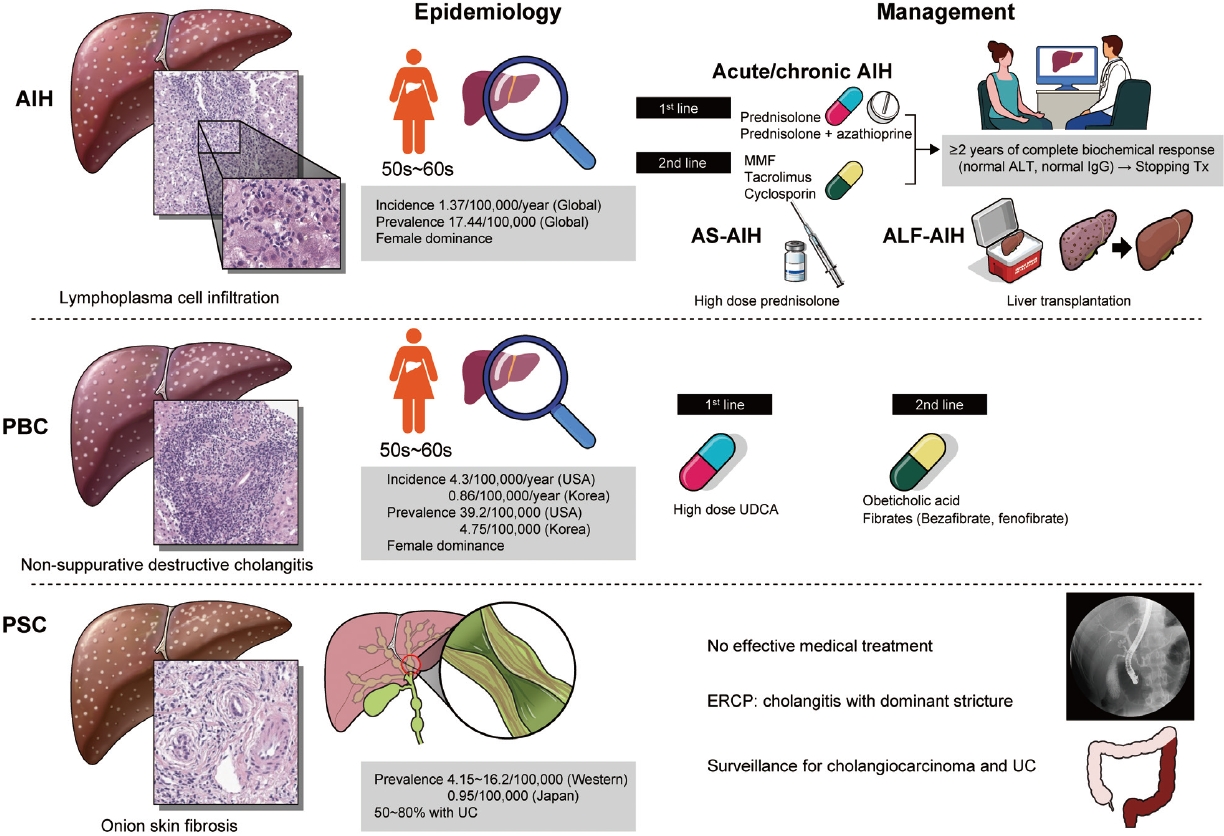
AIH is the hepatocellular damage with the infiltration of lymphocyte and plasma cells, which might be induced by uncontrolled autoreactive CD4 and CD8 T cells. Although AIH might be characterized by elevated serum aminotransferase and immunoglobulin G (IgG), and the detection of autoantibodies, the concept of autoimmunity should be confirmed by the exclusion of other liver diseases [1].
The global annual incidence rate of AIH was 1.37 per 100,000 persons, and similar among the Asian, European, and American populations. The ratio of male to female gender was shown as 1:5. The global prevalence rate of AIH was 17.44 per 100,000 persons with a high level of regional variation [2]. The average age of the onset of AIH is the mid-50s [3].
The strategy of management of AIH is recommended according to disease subtypes such as chronic or acute AIH, acute severe AIH, and acute liver failure (ALF)-AIH [1]. In chronic or acute AIH, glucocorticoid monotherapy or glucocorticoid plus azathioprine could be applied. If remission induction accomplishes, glucocorticoid is tapered to the effective lowest dose or withdrawal, then maintenance therapy with azathioprine ┬▒ low dose glucocorticoid should continue. A recent study suggested that a prolonged complete biochemical response which is defined as normalization of aminotransferase and IgG for at least 2 years might be used to stop treatment without liver biopsy [4]. In case of incomplete biochemical response, treatment failure, or drug intolerance, 2nd line treatment including mycophenolate mofetil, tacrolimus, cyclosporin A, or infliximab could be used. In acute severe AIH, high-dose glucocorticoid monotherapy could be applied without azathioprine due to potential hepatotoxicity. At last, in ALF-AIH with encephalopathy, initial evaluation of liver transplantation (LT) should be considered without anticipation of the response to glucocorticoid [5,6]. While thiopurine methyltransferase activity test is encouraged to avoid azathioprine-related toxicity in European and African descendants, nudix hydrolase 15 variant is associated with the adverse effect of azathioprine in the East Asian population [7].
PBC is a chronic cholestatic autoimmune liver disease characterized by the slowly progressive destruction of small intrahepatic bile ducts. The incidence and prevalence vary according to the region. PBC presented less frequently in Eastern than in Western countries. The annual incidence in USA and Korea was 4.3, and 0.86 per 100,000 persons, respectively. The prevalence in USA and Korea was 39.2, and 4.75 per 100,000 persons, respectively [8-10]. PBC occurs most frequently in women in their 50s and 60s. Diagnosis of PBC is based on the three characteristic findings such as serum alkaline phosphatase elevation, positive anti-mitochondrial antibody, and non-suppurative destructive cholangitis in liver biopsy.
The management of PBC is composed of resolving cholestasis and control of its complications. High-dose ursodeoxycholic acid (UDCA) has been approved as a 1st-line therapy, which has dramatically modified the natural course of PBC. However, about 20ŌĆō30% of patients with PBC showed incomplete response to it. Recently, obeticoholic acid presented approximately 50% of treatment response among the incomplete responders to UDCA, where it received accelerated US Food and Drug Administration approval. However, follow-up phase 3 trial (The Clinical Outcomes with OBeticholic Acid in Liver Treatment [COBALT] study) was terminated early due to feasibility challenge in 2021. Therefore, the availability of obeticholic acid is currently limited. Also, add-on of bezafibrate to UDCA in incomplete responders to UDCA significantly improved liver biochemistry and liver stiffness, and LT-free survival [11,12]. Complications of PBC include fatigue, pruritus, osteoporosis, hyperlipidemia, and Sicca syndrome. It is important to manage them properly to improve the quality of life of the patients [8].
PSC is a chronic cholestatic disease of unknown etiology, which is characterized by multifocal stenosis and destruction of the bile ducts owing to inflammation and fibrosis. The prevalence was 4.15 to 16.2 per 100,000 in Northern Europe and North America, while it was 0.95 per 100,000 in Japan. PSC is male-dominant and 50% to 80% of PSC patients in Western countries have ulcerative colitis [13].
PSC is diagnosed by typical cholangiographic findings and the exclusion of secondary causes. Magnetic resonance cholangiography showed diffuse, multifocal short segment strictures and mild dilatation in the intra- and extrahepatic bile ducts looking like beaded appearance [14]. In contrast to PBC, UDCA did not improve survival, and high-dose UDCA increased the risk of adverse outcomes in patients with PSC [15]. Endoscopic retrograde cholangiopancreatography is indicated to treat the cholangitis with dominant stricture or to take a biopsy when cholangiocarcinoma is suspected. Ultimately, LT should be considered in case of liver failure or decompensated cirrhosis.
In epidemiology, the prevalence and incidence of these rare autoimmune liver diseases presented an increasing trend. However, it is not definite whether this trend reflects a real increase of the disease or improved identification due to better awareness of the physician. In the future, with the effort to find the patients early, it is required to develop more sophisticated treatment approaches individually.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





