


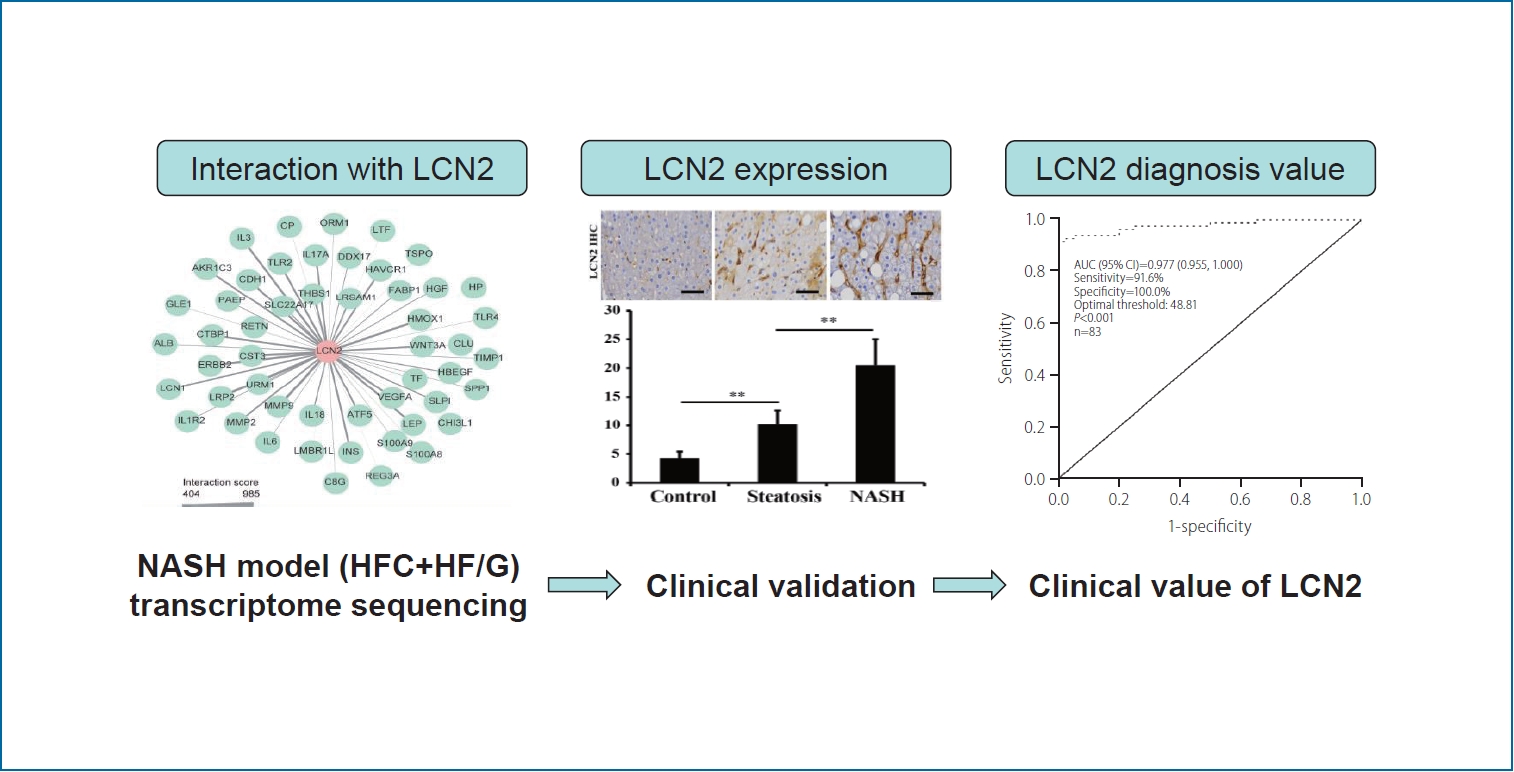
Graphical Abstract
INTRODUCTION
As a leading cause of end-stage liver disease worldwide and the most common liver disorder in western countries, nonalcoholic fatty liver disease (NAFLD) is characterized by nonalcoholic fatty liver (NAFL), nonalcoholic steatohepatitis (NASH), and NASH-associated fibrosis and cirrhosis, affecting around 25% of the general population, and causing a huge burden on society and individuals [1,2]. NASH is an active stage of NAFLD and may progress with fibrosis to cirrhosis and hepatocellular carcinoma, which are the major causes of mortality in NAFLD patients [3]. Lifestyle changes and exercise are the cornerstones for improvement or reversion of NAFL to a normal liver. The clinical diagnosis of NAFLD mainly relies on imaging modalities after exclusion of other etiologies, such as viral infection, drug toxicity, alcohol abuse, or hereditary disorders such as Wilson disease [4]. It is challenging to distinguish NASH from NAFL. The latter often does not need medical intervention. Given that there are only slight to mild abnormalities in NAFLD biochemical tests, ultrasound is less sensitive for fatty content quantitation when a controlled attenuation parameter is used for determination and is often affected by other factors. A more reliable imaging modality is magnetic resonance imaging proton density fat fraction for steatosis quantification. However, the latter is largely used for research purposes and is unavailable for routine examination in regular hospitals. Due to the fact that current NASH diagnosis and progression monitoring strategies are based on the liver histopathology, focusing on steatosis, inflammation, and necroptosis for the determination of the NAFLD activity score (NAS), as well as fibrosis severity (F1ŌĆōF4), liver biopsy still remains an inevitable means for the clinical diagnosis of NASH [5], despite its invasive nature, sampling variations, and discrepancies between histology examiners. Thus, efficient, convenient, and accurate new clinical tests are urgently needed for the clinical diagnosis and progression monitoring of NASH.
Delineating the molecular mechanisms underlying NASH initiation and progression is a breakthrough in identifying novel diagnostic or therapeutic targets for the treatment of NASH. It is known that the dysregulation of certain genes plays important roles in the development of NAFLD or NASH, and novel biomarkers for clinical diagnosis are in high demand [6,7]. Bioinformatics analysis is a commonly used method to screen for potential biomarkers in various diseases. For example, based on bioinformatics analysis, Feng et al. [8] revealed novel core genes closely associated with NAFLD and the progression from NASH to hepatocellular carcinoma. Based on bioinformatics analysis and a text-mining approach, a 25-gene signature was identified to be dysregulated in NASH, and these genes were mainly involved in the transition from steatosis to NASH, which are involved in insulin resistance, inflammation, and lipid metabolism [9]. Chemokine receptor-like 2 is differentially expressed in NASH, and its expression level is positively correlated with NAS and the extent of fibrosis in NASH patients [10]. Therefore, Cenicriviroc is a specific antagonist for targeting CCR2/5 receptor, and is currently under phase III clinical trial for clinical use approval [11].
In fact, the molecular investigation based on mouse NASH models further shortens the process of finding biomarkers in humans [12]. Previous transcriptomic comparisons between different NASH mouse models and NASH patients have shown that high-fat diet feeding-induced NASH is more similar to human NASH compared with other mouse models [13]. In our previous study, a long-term (12 months) feeding high fat/calorie diet plus high fructose/glucose in drinking water (HFCD+HF/G) drinking water recapitulated the pathophysiologic alternations from NAFL, NASH, fibrotic progression/early cirrhosis to premalignant transformation, and provided a full spectrum of metabolic, inflammatory, and fibrotic progression [14]. Transcriptome sequencing was subsequently performed to further investigate potential NASH-related genes and their possible functions in the liver samples of this model. Lipocalin 2 (LCN2), a key gene that was significantly dysregulated in this NASH model, was identified from transcriptome analysis. The increased expression of LCN2 was further confirmed along with the progression of NASH, and serum LCN2 levels were found to be significantly elevated in subjects with steatosis and NASH, compared to that in healthy controls. The findings of this translational study shed new light on novel LCN2 as a valuable NAFLD biomarker, especially for the transition from NAFL to NASH.
MATERIALS AND METHODS
NASH mouse model and experimental design
In total, 30 male C57BL/6J mice (specified pathogen free grade, 25ŌĆō28 g) were purchased from Nanjing Biomedical Research Institute of Nanjing University (Nanjing, China). Mice were fed diet and water ad libitum in a standard feeding environment. All mice were divided into two groups: the HFCD+HF/G group (n=20) and the control group (n=10). Mice in the HFCD+HF/G group were fed a high-fat/calorie diet with 60% kcal derived from fat (cat. no. D12492i, Research Diets, New Brunswick, NJ, USA) and drinking water with 42 g/L D-fructose/D-glucose (55% fructose and 45% glucose) (Sigma-Aldrich, St. Louis, MO, USA) [15]. Meanwhile, mice in the control diet group were given the standard pellet chow diet of 12% kcal from fat, as well as water. All animals were weighed weekly. Mice were sacrificed at 5, 9, and 12 months of feeding via anesthesia with sodium pentobarbital (anesthetic dosage: 60 mg/kg). Blood samples were collected from the orbital sinus, followed by the serum, obtained using centrifugation. Subsequently, all mice were sacrificed via cervical dislocation, and the liver tissue was excised and separated into three parts: 1) formalin fixation and paraffin embedding, 2) total RNA extraction (TRIzol reagent [Ambion, Carlsbad, CA, USA]; 1 mL), and 3) cryopreservation (-80┬░C). The animal experimental protocol was approved by the animal ethics committee of Fudan University School of Basic Medical Sciences, and all experimental procedures were in compliance with the Guidelines for the Care and Use of Laboratory Animals published by the US National Institutes of Health.
Histopathological analysis
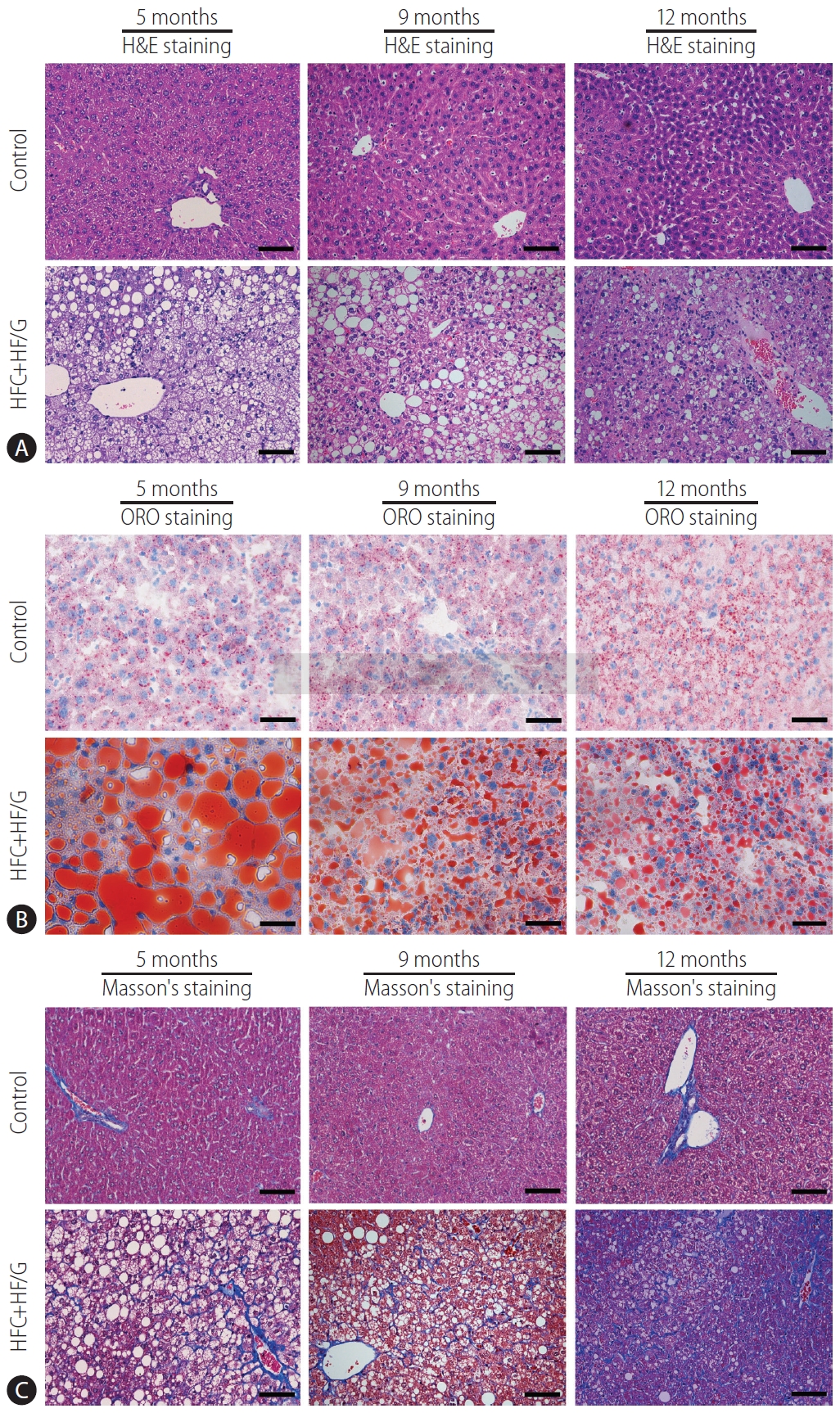
The liver sections from all groups were examined for inflammatory infiltration, necrosis, liver fibrosis, and steatosis. Paraffin-embedded sections were stained using hematoxylin and eosin (H&E) and MassonŌĆÖs trichrome for microscopic examination. Oil red O staining was performed to evaluate hepatic steatosis. In brief, the liver sections were fixed using 60% isopropanol, and stained using 0.3% Oil red O. GillŌĆÖs hematoxylin was used for counterstaining after washing with 60% isopropanol. Finally, after washing with a 4% acetic acid solution, the sections were mounted using an aqueous solution. Sections were visualized at a ├Ś200 magnification (H&E staining) and a ├Ś400 magnification (Oil red O staining) using a Nikon Eclipse E600 microscope (Nikon, Kawasaki, Japan).
RNA-sequencing for transcriptome and bioinformatics analysis
Total RNA from the liver tissues of the three NASH and three control mice at 12 months of feeding was extracted using TRIzol reagent and was examined using a spectrophotometer (Foss NIRSystems 6500; FOSS NIRSystems, Inc., Laurel, MD, USA). Moreover, the NEBNext┬« UltraŌäó DNA Library Prep Kit for Illumina┬« (New England Biolabs, Inc., Ipswich, MA, USA) was used to construct the RNA-seq library. Finally, the Illumina Hiseq 4000 platform (PE150, San Diego, CA, USA) was used for sequencing data collection. The fragments per kilobase million data from the transcriptome sequencing samples were used as the original data. Sample quality and read numbers were assessed using FASTQC (version 0.11.5; Babraham Institute, Cambridge, UK) [16]. Clean reads were obtained by filtering the joint and low-quality sequences from all the generated raw data. Subsequently, clean reads were aligned to the reference genome using default parameters. Differential expression analysis of the genes between the HFCD+HF/G and the control diet groups was performed using DEseq [17], and the differentially expressed genes (DEGs) with a |log2-fold change (FC)| >2 and a P<0.05 were screened. Heatmap (1.0.12) and ggplot2 (3.2.1) were used to plot the heat map and volcano plot, respectively. Functional enrichment analysis was performed using ClusterProfiler (3.12.0) [18] using the R package to analyze the DEGs related to Gene Ontology functional terms. Protein-protein interaction (PPI) were retrieved from Search Tool for the Retrieval of Interacting Genes (STRING, http://string-db.org) database [19] with a combined score >0.4, and the PPI network was visualized using Cytoscape software (http://cytoscape.org/) [20].
Quantitative reverse transcription polymerase chain reaction (qRT-PCR) assay
Total liver RNA was reverse transcribed to cDNA using the Prime-Script RT reagent kit (cat. no. RR037A; Takara, Tokyo, Japan). Power SYBR green PCR Master Mix (applied Biosystems, Foster City, CA, USA) using an Eppendorf realplex cycler (Eppendorf International, Hamburg, Germany) was used for the amplification of qRT-PCR and performed in triplicates. Human GAPDH was used as the internal reference gene (cat No: b661304, SANGON Biotechnology Co., Ltd., Shanghai, China), and the remaining primers were designed according to the primer 3.0 online tool and synthesized by SANGON Biotechnology (Supplementary Table 1). The PCR program was performed under the following cycling conditions: 95°C for 10 minutes (activation of Taq enzyme), 40 cycles of 95°C for 15 seconds, and 60°C for 1 minute. The relative expression of genes was calculated using the 2-ΔΔCt method.
Western blot analysis
Protein extraction was performed using RIPA Lysis Buffer, followed by quantitation using a bicinchoninic acid assay kit (Thermo Scientific, Rockford, IL, USA). Denatured protein (40 ╬╝g) was separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred onto a polyvinylidene fluoride membrane (Millipore, Billerica, MA, USA), and then blocked in 5% non-fat milk (with a Tris-HCl-buffered saline solution) for 1.5 hours. After incubation overnight (4┬░C) with the primary antibody (cat no: ab187370, 1:300; Abcam, Cambridge, MA, USA), the SAB goat anti-rabbit IgG secondary antibody (horseradish peroxidase [HRP] conjugated, cat no: L3012-2, 1:6,000) was used for all samples at room temperature for 1 hour. Finally, all results were visualized using the enhanced chemiluminescence kit (Millipore) and a chemiluminescent imaging system.
Immunohistochemical staining
The liver sections were deparaffinized, rehydrated, and pretreated with citrate buffer (10 mM sodium citrate; pH 6.0) to unmask the antigen. After washing with phosphate buffered saline (PBS), sections were incubated with H2O2 (0.3%) for 30 minutes to block the endogenous peroxidase activity and then blocked with 3% bovine serum albumin at room temperature to avoid nonspecific binding of the primary antibody, followed by incubation with a primary anti-LCN2 antibody (1:300; cat No: ab187370; Abcam) at 4┬░C overnight. After washing with PBS three times, sections were incubated with the HRP-conjugated secondary antibody for 2 hours at room temperature. 3,3-Diaminobenzidine chromogenic reagent was added and the sections were incubated for 2 hours at room temperature. Sections were then counterstained with hematoxylin, rinsed, and air-dried. After sealing with a neutral resin, the sections were examined under a fluorescent microscope (TE2000-E; Nikon Corporation, Tokyo, Japan). Ten randomly chosen fields were photographed under a ├Ś400 magnification and were analyzed using ImageJ software (National Institutes of Health).
Patient demographic information
Patients (n=360, aged >18 years) who were diagnosed with NAFLD, including 83 cases of steatosis and 277 cases of NASH, were continuously recruited from the First Affiliated Hospital of Wenzhou Medical University. NASH patients were diagnosed based on the practice guidelines from the American Association for the Study of Liver Diseases (AASLD) [21]. In the AASLD guidelines, NASH is defined as the presence of >5% hepatic steatosis with inflammation and hepatocyte ballooning degeneration, with or without any fibrosis. Nonalcoholic hepatic steatosis, also known as simple steatosis (NAFL), is an early manifestation of NAFLD, in which macrovesicular or predominant macrovesicular steatosis involves more than 5% of hepatocytes and may be accompanied by mild nonspecific inflammation [22]. All patients with NAFLD were pathologically confirmed using the liver biopsy. Patients who met the following criteria were excluded: 1) alcohol consumption of more than 20 g/day; 2) patients with viral hepatitis B or C, autoimmune hepatitis, primary biliary cholangitis, or WilsonŌĆÖs disease; 3) long time consumption of nonsteroidal antiinflammatory drugs, calcium antagonists, tamoxifen, amiodarone, corticosterone, isoniazid, or methotrexate; 4) pregnant or lactating women; and 5) patients with liver cancer or other benign/malignant tumors. Additionally, a total of 40 healthy individuals and 59 cases of alcoholic fatty liver disease (AFLD) patients were recruited for additional control and verification. All recruited individuals agreed to participate in the study and provided written informed consent for the liver biopsy, collection of clinical samples, and other information. The study was approved by the Ethics Committee of the First Affiliated Hospital of Wenzhou Medical University (no. 20190023, vote: 32/38).
Measurement of serum and urine LCN2 and other clinical parameters
Blood (2,000 rpm) and urine samples (1,000 rpm) were centrifuged for 10 minutes. The supernatant was collected, and serum LCN2 and urine LCN2 levels were measured using an up-converting phosphor-technology-based lateral flow assay. Body mass index (BMI) was calculated as weight (kg)/height in meters squared. The serum levels of total cholesterol, total bilirubin, high-density lipoprotein (HDL) cholesterol, low-density lipoprotein (LDL) cholesterol, alanine aminotransferase (ALT), aspartate aminotransferase (AST), and ╬│-glutamyltransferase (╬│-GT) were detected using an automated analyzer (AU680, Beckman Coulter, Chaska, MN, USA) using standard methods.
Statistical analysis
SPSS ver. 19.0 (SPSS Inc., Chicago, IL, USA) software was used to perform all statistical analyses in the current study. Quantitative data were assessed for normality using the Shapiro-Wilk test. If continuous variables were conformed to a normal distribution, data were expressed as mean┬▒standard deviation, followed by analysis of the difference using a t test (between two groups) and one-way analysis of variance (among more than two groups); if not, data were presented as the median (interquartile range) and their differences were compared using the Mann-Whitney U test (between two groups) and Kruskal-Wallis (H) test (among more than two groups). A P<0.05 was considered statistically significant. Qualitative data were expressed in the form of number (%), and the differences between groups were compared using the chi-square test. Univariate logistic regression analysis was performed on the variables with significant differences compared with the baseline information, and variables with statistical significance in univariate logistic regression were further included in the multivariate stepwise regression model. The diagnostic value of potential NASH-related genes for NAFLD (steatosis and NASH) was evaluated via analysis of the area under the receiver operating characteristic (ROC) curve (AUC), specificity, and sensitivity. A two-sided P<0.05 was selected as the cut-off value for significant analysis.
RESULTS
Mouse NASH model establishment
After 12 months of HFCD+HF/G feeding, a NASH model with severe fibrosis was successfully established (Fig. 1). H&E staining indicated that HFCD+HF/G-fed mice showed inflammatory cell infiltration, focal necrosis in the leaflets, ballooning cells and binuclear cells, and fibrosis (Fig. 1A). Oil red O staining showed extensive red lipid droplet deposition in the cytoplasm of HFCD+HF/G-fed mice, suggesting that mice in the HFCD+HF/G group exhibited severe hepatic steatosis (Fig. 1B). Moreover, MassonŌĆÖs staining showed progressive fibrosis with characteristics of early cirrhosis in mice in the HFCD+HF/G group (Fig. 1C). In addition, our previous studies have verified that feeding HFC diet plus HF/G resulted in severe obesity, impaired liver function, glucose intolerance, insulin resistance and steatohepatitis with significant necroptotic inflammation [14].
Analysis of RNA-sequencing transcriptome data
In order to investigate the potential molecular mechanisms underlying NASH, RNA-sequencing was performed. In total, 504 genes were differentially expressed in HFCD+HF/G-fed mice in comparison with control diet-fed mice, of which 328 genes were upregulated, whereas 176 were down-regulated (Supplementary Data 1). The heatmap of DEGs indicated that they could obviously distinguish HFCD+HF/G-fed mice from control diet-fed mice (Fig. 2A). The volcano plot revealed that DEGs were significantly divided into upregulated and downregulated genes (Fig. 2B). These DEGs were significantly enriched in different biological processes such as inflammation, including leukocyte migration, cell/leukocyte chemotaxis, leukocyte cell-cell adhesion, positive regulation of defense response, and tumor necrosis factor superfamily cytokine production (Fig. 2C). These results suggested that HFCD+HF/G-fed mice might possess more inflammatory responses than that in control diet-fed mice.
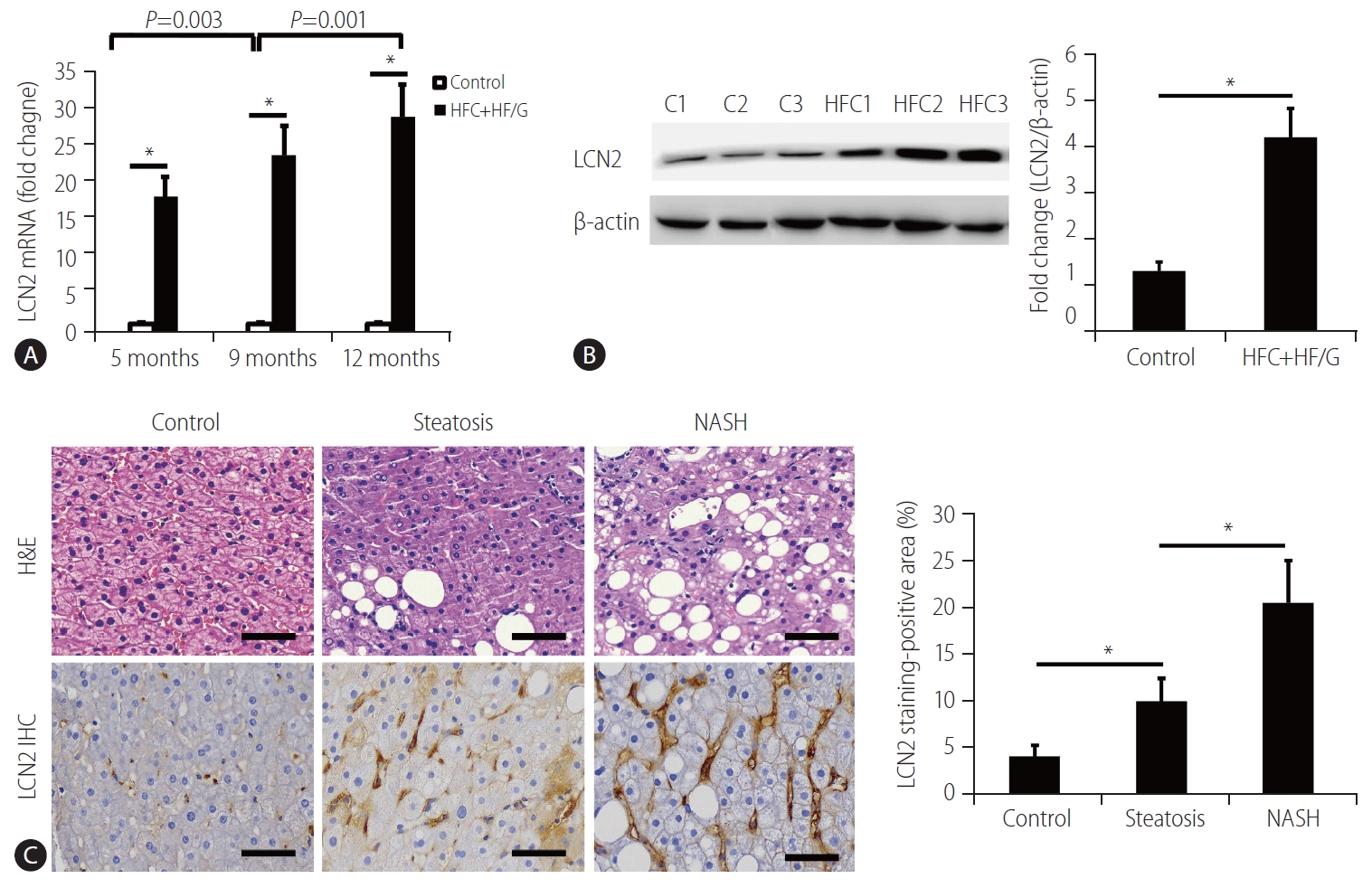
LCN2 encodes a neutrophil gelatinase-associated lipocalin that functions in innate immunity and has recently been reported as a novel player in inflammation [23]. In the current study, LCN2 expression was significantly upregulated (fold change=28.60; log2-FC=4.84; P=0.000226) in HFCD+HF/G-fed mice. Further, the interactions between LCN2 and other genes were screened using the STRING database, and the PPI network is shown in Figure 2D. It appears that LCN2 may interact with various interleukins (such as interleukin [IL]1R2, IL3, IL6, IL18, and IL17A), leptin, cadherin 1, LDL receptor related protein 2, and toll-like receptors (TLR2 and TLR4), or may be involved in the cascades of these inflammatory responses. Therefore, LCN2 might be a key molecule in the inflammatory and immune responses during NASH development and progression, although details of its molecular interplay remain to be elucidated.
Validation of LCN2 expression in NASH mouse model
The upregulated LCN2 expression was confirmed in the liver tissues of NASH mice using both qRT-PCR and Western blotting analysis (Fig. 3A, B). Compared to the control diet-fed mice, LCN2 expression at the mRNA level significantly increased in HFCD+HF/G-fed mice, and there was a statistically significant increase in LCN2 expression from 5 to 12 months (Fig. 3A). LCN2 expression at the protein level was upregulated in NASH mice at the time of sacrifice, compared to that in control mice (Fig. 3B). This was consistent with the results from the transcriptome data.
Measurement of serum LCN2 levels in different groups of patients
In the present study, 360 NAFLD patients, 40 healthy individuals, and 59 AFLD patients were enrolled. Based on the definitions for both steatosis and NASH described in the methods, the NAFLD patients were divided into steatosis (n=83) and NASH groups (n=277), and representative micrographs of their liver sections stained with H&E are shown in Figure 3C. LCN2 expression in the liver tissues of patients with steatosis and NASH was also determined using immunohistochemical staining. Morphometrically, LCN2 stained-positive area was significantly increased in both steatosis and NASH groups, compared to that in healthy individuals (Fig. 3C).
There was a significant elevation in serum LCN2 levels in both steatosis and NASH groups compared to healthy individuals (controls), and NASH patients had the highest serum LCN2 levels among these groups (Fig. 4A), suggesting that the serum LCN2 levels might be elevated along with the NAS and pathological parameters, such as steatosis, ballooning degeneration, intralobular inflammation and fibrosis. There was no significant difference in the serum LCN2 levels between the NAFLD and AFLD groups (Fig. 4B). Moreover, there was no significant difference in urinary LCN2 levels between NAFLD and healthy individuals (Fig. 4C).
Multivariate analysis of serum LCN2 levels with other parameters
The demographic information of 360 NAFLD patients (including 83 steatosis and 277 NASH patients) and 40 healthy individuals is shown in Table 1. There were significant differences in the serum LCN2 levels and in multiple clinical characteristics between NAFLD patients and healthy individuals. Of note, there were no significant differences in total bilirubin, direct bilirubin, indirect bilirubin, HDL cholesterol, LDL cholesterol, alkaline phosphatase, glutamyl transferase and glucose between NASH and steatosis patients. NASH patients exhibited significant differences from steatosis patients in many biochemical, metabolic, and pathologic variables, such as serum LCN2 level, total cholesterol, total protein, albumin, ALT, AST, AST/ALT, glutamyl transferase, urea nitrogen, fasting insulin, steatosis, ballooning change, intralobular inflammation, and fibrosis extent (Table 1).
The variables with statistical significance between NAFLD and healthy individuals in Table 1 were included in the univariate logistic regression analysis. The results showed that serum LCN2, BMI, weight, total cholesterol, triglycerides, LDL cholesterol, HDL cholesterol, ALT, AST, AST/ALT, glutamyl transferase, glucose, fasting insulin and indirect bilirubin were all associated with NAFLD (all P<0.05, Table 2). Furthermore, these variables were included in the multivariate stepwise regression model. It is intriguing that three of them, including serum LCN2 level (P<0.001), BMI (P=0.001), and LDL cholesterol (P=0.002), were positively correlated with NAFLD (Table 2). The area under the ROC curve (AUC) of serum LCN2 level for diagnosis of NASH was 0.987 with a specificity of 100% and a sensitivity of 93.5% (Fig. 4D). According to the ROC curve, the optimal cut-off value of LCN2 for the diagnosis of NASH was Ōēź49.205 g/mL (Supplementary Data 2). The false positive and false negative rates of serum LCN2 level for the diagnosis of NASH were 0% and 6.5%, respectively. The area under the ROC curve (AUC) of serum LCN2 level was 0.977 with a specificity of 100% and a sensitivity of 91.6% for the diagnosis of steatosis (Fig. 4E). According to the ROC curve, the optimal cutoff value for diagnosis of steatosis was Ōēź48.81 ng/mL (Supplementary Data 2). The false positive and false negative rates of serum LCN2 level for the diagnosis of steatosis were 0% and 8.4%, respectively. The low false positive and false negative rates confirmed the high diagnostic value of serum LCN2 in both NASH and steatosis.
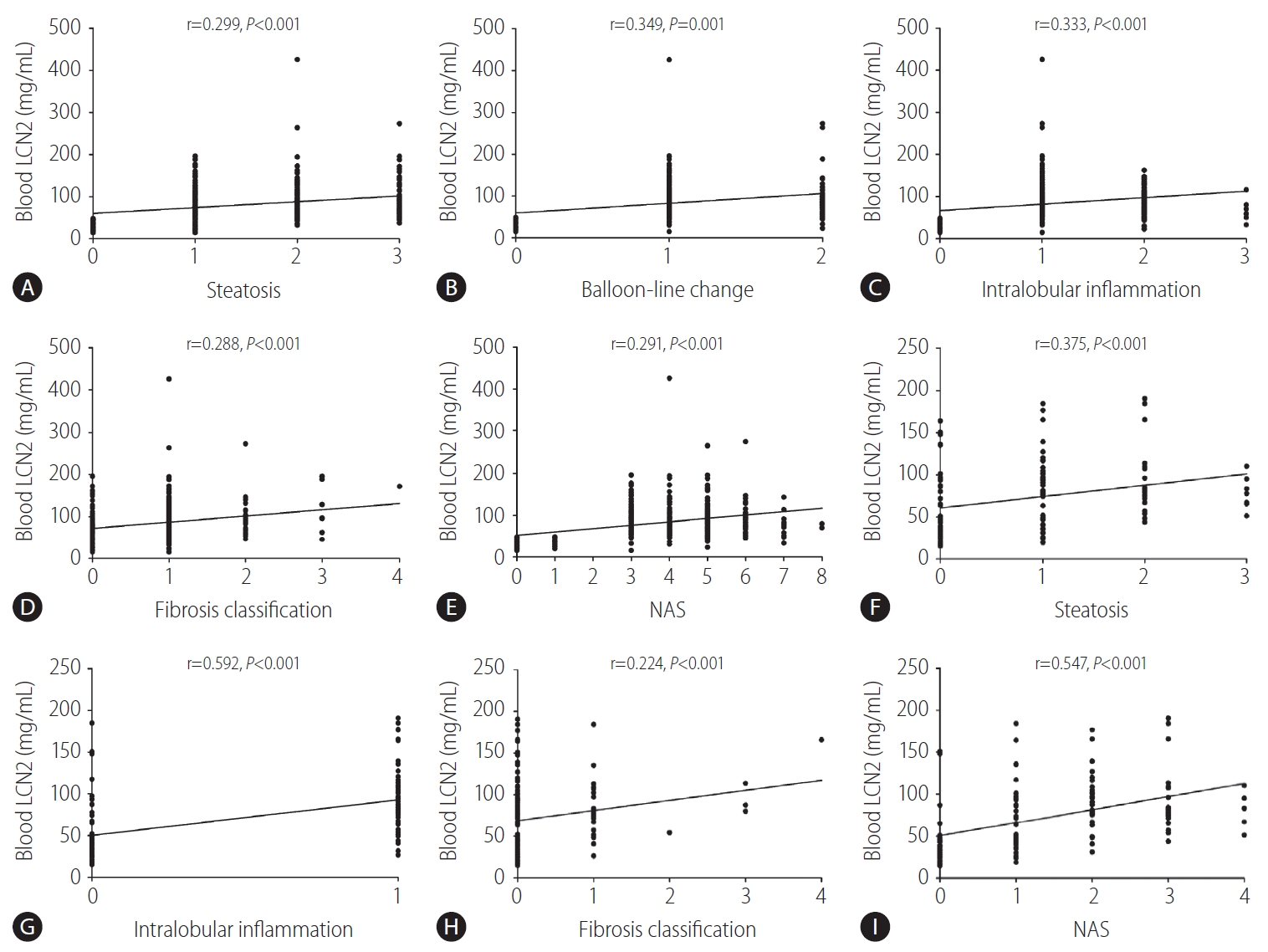
Furthermore, correlation coefficient analysis was performed to determine whether the serum LCN2 levels alter along with the pathologic components of NASH diagnosis in both NASH and steatosis groups. In the NASH group, the serum LCN2 levels showed moderate positive correlations with steatosis, balloon-like change, intralobular inflammation, fibrosis classification and the NAS score (Fig. 5A-E). Similar results were observed in the steatosis group, in which the serum LCN2 levels were positively correlated with steatosis, intralobular inflammation, fibrosis classification and the NAS score (Fig. 5F-I). These data indicate that the serum LCN2 level might be a valuable biomarker that could potentially reflect the transition from NAFL to NASH and fibrotic progression.
DISCUSSION
NASH is a progressive stage of NAFLD characterized by liver steatosis, inflammation, hepatocellular necroptosis and different extent of fibrosis [24]. Elucidation of the mechanism by which inflammation is triggered and contributes to the transition from NAFL to NASH is a major issue in clinical practice. Through long-term HFCD+HF/G feeding, a unique murine model of pathophysiologic transition from NAFL to NASH, and further fibrotic progression was re-established [14]. Through transcriptome analysis of a significantly differential gene pool, LCN2 was identified to be elevated over time during this transition and progression. Its association with inflammatory and innate immunity (e.g., leukocyte migration and cell/leukocyte chemotaxis) is well indicated by complex interactions with various interleukins (e.g., IL-6 and IL-17A) and toll-like receptors (TLR-2 and TLR-4). Moreover, the serum LCN2 levels significantly increased in NAFLD patients and correlated with steatosis, intralobular inflammation, fibrosis extent and NAS in the present study.
LCN2 encodes a neutrophil gelatinase-associated lipocalin that functions in innate immunity and is thought to be a novel player in inflammation [23]. Ye et al. [25] showed that elevated circulating LCN2 levels in NASH patients and mouse models were correlated with increased neutrophil infiltration and aggravated steatohepatitis by facilitating the neutrophil-macrophage crosstalk by inducing chemokine receptor CXCR2. Elevated LCN2 levels have been found to be correlated with hepatic inflammation in NAFLD patients [26]. However, extensive studies are unavailable to determine how LCN2 levels alter along with the transition from NAFL to NASH, and how it further progresses to severe fibrosis in long-term lipotoxicity conditions. The histopathologic and RNA-seq findings from our well-characterized HFCD+HF/G-fed mice revealed that LCN2 might be involved in inflammatory and innate immunity functions through interaction with various interleukins (e.g., IL6 and IL17A) and toll-like receptors (TLR2 and TLR4). TLRs are highly conserved proteins that function in the activation of innate immunity [27]. TLR signaling is activated in NAFLD, resulting in an increase in proinflammatory cytokines and other downstream molecules [28]. Arrese et al. [29] reported that innate immune activation was considered a major factor in triggering and aggravating hepatic inflammation in the transition from NAFL to NASH. Moreover, resident Kupffer cells and microphages from circulation that respond to liver injury could release cytokines, chemokines and other pro-inflammatory signals, which promote hepatocellular necroptosis; in turn, injured hepatocytes release damage-related molecules to further amplify the inflammatory responses by activating the innate immune system [30,31]. Therefore, it is speculated that LCN2 might play an important role in the transition from NAFL to NASH by activating innate immunity, which in turn facilitates inflammatory responses. Physiologically as a lipocalin, LCN2 transports small hydrophobic molecules, such as fatty acids and steroids [32]. LCN2 was found to be associated with steatosis by mediating lipid homeostasis [33]. Growing evidence has demonstrated that an increased LCN2 expression in adipose tissue may prevent functional deterioration of adipocytes due to aging, liver lipid accumulation, and steatosis [34]. However, published investigations are mostly at a pilot scale, and no series of investigations are available to determine whether serum LCN2 is able to distinguish healthy individuals from NAFL or NASH patients. In the present study with a sufficient patient number in each group, serum LCN2 level significantly increased in NAFLD (NASH and steatosis) patients, and correlated with steatosis, intralobular inflammation, and the extent of fibrosis. Therefore, we speculate that LCN2 may be involved in NAFLD development by affecting lipid homeostasis and the formation of hepatic inflammation and steatosis.
Liver biopsy is an invasive method used for the diagnosis of liver diseases, and bears sampling difference and discrepancy in histological evaluation [35]. However, it is required for pathologic determination of the progression and regression of NASH or fibrosis when pharmacotherapeutic interventions are considered [36]. As demonstrated in the demographic data of the present study, biochemical tests did not show dramatic differences between NAFL and NASH. In fact, there is no reliable surrogate marker available so far to facilitate the monitoring of the progression from NAFL to NASH, or regression due to lifestyle changes or pharmacological invention. Therefore, it is of great significance to develop noninvasive and valuable biomarkers for the clinical diagnosis of NAFLD. Previous studies have indicated that the serum LCN2 level may be used as a diagnostic marker for various diseases, including colorectal cancer and cholangiocarcinoma [37,38]. However, based on a rat model that was induced via a fructose-enriched diet, an elevated LCN2 level was suggested to be a biomarker for fructose-induced fatty liver [39], which is supported by the fact that LCN2 expression is indicative of pathological conditions, including hepatic steatosis and inflammation [40]. Largely in consistence, our animal model of well-characterized NASH with fibrosis progression indicates that the liver LCN2 was increased along with the transition from NAFL to NASH and further fibrotic progression. More convincingly, the LCN2 levels were progressively elevated, along with the transition from healthy individuals to NAFL, and further progression to NASH in this observation, in which all NAFLD subjects were pathologically confirmed. The AUC of the serum LCN2 level was 0.987 with a specificity of 100% and a sensitivity of 93.5% for the diagnosis of NASH, and was 0.977 with a specificity of 100% and a sensitivity of 91.6% for the diagnosis of steatosis. The false positive and false negative rates of serum LCN2 for the diagnosis of NASH were 0% and 6.5%, respectively, and those for diagnosis of steatosis were 0% and 8.4%, respectively. Given a high diagnostic value of serum LCN2 levels, it is promising that the serum LCN2 level might be a valuable biomarker for the clinical diagnosis of patients with NAFL and NASH progression.
Notably, BMI and LDL cholesterol were risk factors of NAFLD as suggested by the results of multivariate logistic regression analysis in the present study. A community-based retrospective longitudinal cohort study revealed that BMI is the most useful predictive factor for the onset of NAFLD [41]. Min et al. [42] demonstrated that a dysregulation of the cholesterol metabolism is correlated with the severity of NAFLD. Furthermore, correlation analysis demonstrated that the serum LCN2 levels were positively correlated with multiple clinical indicators, such as ALT, creatinine, fasting insulin, steatosis, intralobular inflammation, fibrosis grade and NAS; however they were negatively correlated with HDL cholesterol. The serum ALT value is widely used as a surrogate marker of liver injury, however patients with NAFLD may have normal ALT values but display significant steatohepatitis and advanced fibrosis [43]. A lower ratio of creatinine to body weight was confirmed to be a predictor of the incidence of NAFLD [44,45]. A simple clinical scoring system, including fasting insulin, has been shown to have sufficient accuracy and simplicity in predicting NASH in NAFLD patients [46]. Steatosis, intralobular inflammation, fibrosis grade, and NAS are common indicators of the severity of NAFLD. Although the relationship between the serum LCN2 level and these clinical indicators has not been fully clarified, based on their correlation and the value of these clinical indicators in NAFLD, we speculate that the combination of serum LCN2 level with these clinical indicators will better predict the onset and severity of NAFLD.
As most clinical studies, there were some limitations in this study. First, it lacks a case-control observation in a large cohort size to verify the value of LCN2 in the diagnosis of NASH. Second, the exact biological function of LCN2 and the intersections between LCN2 and other key genes should be further investigated using corresponding functional experiments. Last, although the serum LCN2 levels were compared between NAFL and NASH groups, ROC curve analysis was unable to establish an optimal cut-off value of serum LCN2 levels for distinguishing NASH from NAFL subgroups. Therefore, well-designed and larger-scale studies are needed to confirm our findings.
In conclusion, the mouse NASH model caused by HFCD+HF/G feeding recapitulates the pathological changes (inflammatory infiltration, hepatic steatosis, and fibrosis) and gene expression pattern changes over time. LCN2 was identified from dysregulated genes and was implicated to be associated with inflammation. It was highly expressed in the livers of NASH patients and elevated in patients in the transition from NAFL to NASH, and correlated with a number of biochemical, metabolic and pathologic parameters of NASH initiation and progression. Therefore, LCN2 may serve as a valuable biomarker for monitoring the initiation and progression of NASH.


 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Supplement1
Supplement1 Print
Print





