


FOCAL NODULAR HYPERPLASIA (FNH)
Introduction
Focal nodular hyperplasia (FNH) accounts for up to 8% of all liver neoplasms, with an estimated prevalence of 0.9%[1]. FNH is most often found in women (80% of cases) of reproductive age. The majority of FNH is solitary (80%), usually smaller than 5 cm, and occurs near the liver surface [2]. It may also be multiple (multiple FNH syndrome) or associated to other benign nodules, such as hemangioma or adenoma. Clinical course is asymptomatic in the slightly majority of cases, as the lesion is often discovered incidentally or as a palpable abdominal mass on physical examination. The widespread use of both ultrasounds (US) and computed tomography (CT) in the clinical practice has increased the detection rate of FNH. Although an association with oral contraceptive use has been speculated, owing to increased prevalence of these tumors in young women, studies have shown that FNH is not hormonally dependent nor affected by oral contraceptives or pregnancy [3]. Definitive diagnosis of FNH is obtained by imaging in the majority of cases, particularly in those exhibiting a central scar at contrast CT and magnetic resonance imaging (MRI), but radiologically atypical cases occur and require the use of the liver biopsy [2]. Angiography typically demonstrates a hypervascular mass with enlarged peripheral vessels and a single central feeding artery. This so-called ŌĆ£wheel-spokeŌĆØ appearance with the vessels radiating out from the center of the tumor may help distinguish FNH from hepatic adenoma.
Pathology
FNH is thought to occur as a result of a hyperplastic response to a vascular anomaly, with ensuing disorganized growth of hepatocytes and bile ducts. In a normal liver, the artery within the portal tract supplies the peribiliary vascular plexus, the portal vein wall, and the portal tract interstitium [4,5]. FNH results from portal tract injury (due either to portal tract inflammation or arterial ischemia) leading to arterio-portal or hepatic venous shunts, arterialized sinusoids with hepatocellular hyperplasia, and, in some cases, cholestasis. Portal tract remodeling leads to ductular reaction and porto-periportal fibrosis. Arterial hyperperfusion (and resultant hyperoxemia) leads to increased expression of vascular endothelial and somatic growth factors and activation of hepatic stellate cells [6]. The latter are thought to be responsible for the formation of the characteristic central scar [7].
FNH and FNH-like lesions also occur in association with other vascular abnormalities, such as hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber disease) and congenital absence of the portal vein [8]. In Budd Chiari syndrome and in rarer vascular disorders, FNH and large regenerative nodules/FNH-like have also been reported [9]. The latter lesions can also be encountered in the context of cirrhosis as a result of a locoregional abnormal vascular supply. In vascular disorders, FNH has to be distinguished by hepatocellular adenoma, especially of telangiectatic type and other mimickers [9]. Oxaliplatin-based chemotherapy is also known to be associated with liver vascular injury, sinusoidal dilatation and formation of nodular regenerative hyperplasia, a process characterized by nodular liver remodeling [10,11].
Macroscopically, FNH is a pale, firm lesion distinct from the surrounding liver. Histologically, FNH is sharply demarcated from the normal liver but generally lacks a true capsule. The architecture of the lesion suggests a regenerative rather than a neoplastic process; as such, the lesion in a limited sample, can be difficult to distinguish histopathologically from cirrhosis with regenerating nodules. This difficulty can be readily overcome by informing the pathologist of the diagnostic imaging findings and providing a sample of normal-appearing liver tissue, away from the mass. Unlike hepatic adenoma, bile duct hyperplasia, ductular reaction and KupfferŌĆÖs cells are prominent. The central portion of a FNH is usually composed of fibrous septa containing a sparse inflammatory infiltrate and prominent thick-walled, abnormal blood vessels, which may give the appearance of a scar on imaging. This finding, however, is by no means constant or pathognomonic. Around 8% of FNH have a pseudocapsule [12-14]. FNH can be divided into classical (80%) and non-classical or atypical (showing unusual features such as steatosis, large cell changes, Mallory bodies, cholestasis) [15]. The abnormal architecture or vascular malformations may be absent in non-classical forms, but bile duct proliferation is always present [16].
Molecular features
FNH is a nodular polyclonal tumor-like lesion, that does not undergo hemorrhage or malignant transformation. Clonal analysis using the HUMARA test demonstrated the reactive polyclonal nature of liver cells in FNH in 50-100% of the cases [17]. Messenger RNA (mRNA) expression levels of the angiopoietin genes (ANGPT1 and ANGPT2) involved in vessel maturation are altered, with the ANGPT1/ANGPT2 ratio increased as compared with normal liver, cirrhosis, and other liver tumors [18]. These data support the importance of vascular alterations in the pathogenesis of FNH.
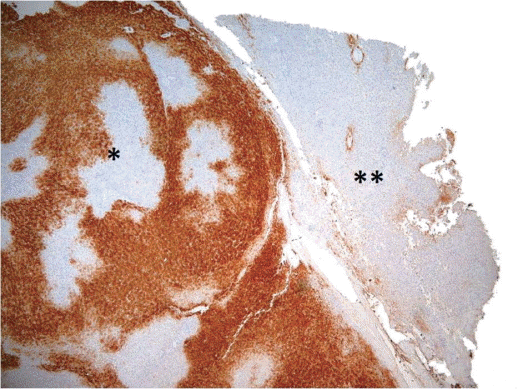
Non-clonal ╬▓-Catenin activation, without mutations, has been shown to occur in FNH, contributing to hepatocellular hyperplasia and regeneration [19]. ╬▓-Catenin appears to be activated due to perivascular hypoxic conditions, modulating cellular regeneration and hyperplasia. The molecular mechanisms of this activation are uncertain, but mutations in ╬▓-catenin or Axin1 have never been shown. Consequently, normal ╬▓-catenin membrane pattern of immunoreactivity can be demonstrated in FNH. The activation of ╬▓-catenin explains the expansion of hepatocytes expressing glutamine synthetase (GS), a target gene of ╬▓ -catenin that is useful for the histologic diagnosis [17,19]. A map-like perivascular pattern of staining for GS is usually seen and it is different by the one exhibited by the adjacent liver parenchyma (Fig. 1). Nevertheless, pathologists should be aware that atypical patterns of staining exist [20]. Notably, FNH-like lesions arising in cirrhosis do not usually display this pattern of staining.
Treatment
As with all benign hepatic tumors, symptoms and the inability to exclude malignancy are the most common reasons for resection of FNH. In some cases, diagnostic uncertainty prompts a percutaneous needle biopsy under US control [21]. When a firm histologic diagnosis is not available, surgical resection with a rim of normal tissue is required. When the diagnosis of FNH is made by imaging, patients usually undergo a regular follow-up. Because FNH can occur in association with hepatocellular adenoma, any change in size, number, or symptoms of the putative FNH should prompt reconsideration of surgical resection.
HEPATOCELLULAR ADENOMA (HCA)
Introduction
Hepatocellular adenoma (HCA) is an uncommon neoplasm of the liver described in young women of child-bearing age with history of estrogen-based, oral contraceptive steroid (OCS) use, and less frequently reported in men. HCA is usually solitary, but a specific clinical entity, liver adenomatosis, is defined as the occurrence of more than 10 HCAs. The detection of HCA is usually incidental, with imaging performed for non-hepatic indications, and has increased with the implementation of new, more specific, imaging modalities. In fertile women, the estimated annual incidence is 3 to 4 per 100,000 per year in North America and Europe [22-24] but not in Asia, likely for the lesser use of OCS [25,26]. Increasing evidence also suggests obesity and metabolic syndrome as emerging risk factors for HCA [23]. One potential mechanism linking obesity to HCA involves activation of the IL- 6 signaling pathway and the inflammatory variant of HCAs. HCA is also reported in patients affected by other metabolic disturbances, including glycogen storage disease type I, with an increased frequency of malignant transformation [27,28]. A minority of HCA are documented in male patients, which suggests male sex also be a condition at risk for malignant transformation and generally reported to be associated with androgen assumption or metabolic disturbances [29]. In fact, ŌĆ£spontaneousŌĆØ adenoma, defined as HCA in absence of any risk factor, are a rare entity.
Large tumors can become symptomatic if they rupture or bleed spontaneously, leading to hemorrhagic shock [30]. Besides the risk of rupture and bleeding, a small subset of HCA has the potential to undergo malignant transformation, but HCC developing from this route are thought to behave less aggressively than those arising in other settings [31]. Hence, it is important to correctly diagnose and treat HCA lesions.
Pathology
HCA is a typically nodular lesion with size ranging from microscopic foci up to 20 cm in diameter. On cut section, the tumor parenchyma is soft and relatively uniform, although areas of congestion, necrosis, hemorrhage, or fibrosis can be observed. The margins of the lesion are ill-defined, without fibrous capsule. In livers with adenomatosis, there may be hundreds of lesions visible as minute ill-defined nodules. HCA may be similar in color and texture to the background liver, but are more easily seen when there is lesional steatosis, major congestion and hemorrhage or degenerative changes. The background liver is usually normal, though there may be pallor, fibrosis, or brown pigmentation related respectively to fatty liver disease, glycogen storage disease, iron overload, or rare vascular disorders [8].
HCA is defined as tumoral monoclonal proliferation of well-differentiated, usually bland-looking, hepatocytes arranged in sheets and cords that are usually one, or at most two, cells in width. Notably, they are also mostly characterized by the absence of a portal triad and interlobular bile ducts, with these features being of great aid for the diagnosis and distinction from the normal adjacent liver [20]. Tumor hepatocytes have cytoplasms that may be normal, clear (glycogen-rich), steatotic, or contain pigment in lysosomes. Nuclear atypia and mitoses are unusual but may be seen in specific variants [32]. As for FNH it is recommended to sample specimens at the border of HCA, in order to compare morphophenotypical features of intra- and extra- lesional tissue and to sample different areas of the tumor if the latter looks heterogeneous.
Molecular features and classification
The recent molecular classification of HCA in several subclasses revealed the heterogeneity of the disease, refining the understanding of the oncogenic mechanisms activated in benign liver tumorigenesis [33,34]. These studies produced a molecular classification of HCA with a strong translational impact in term of diagnostic immunocytochemical markers, worldwide validated [35-40]. According to this classification HCAs are divided into 4 major subgroups.
Hepatocyte nuclear factor 1A (HNF1A) inactivated HCA (H-HCA)
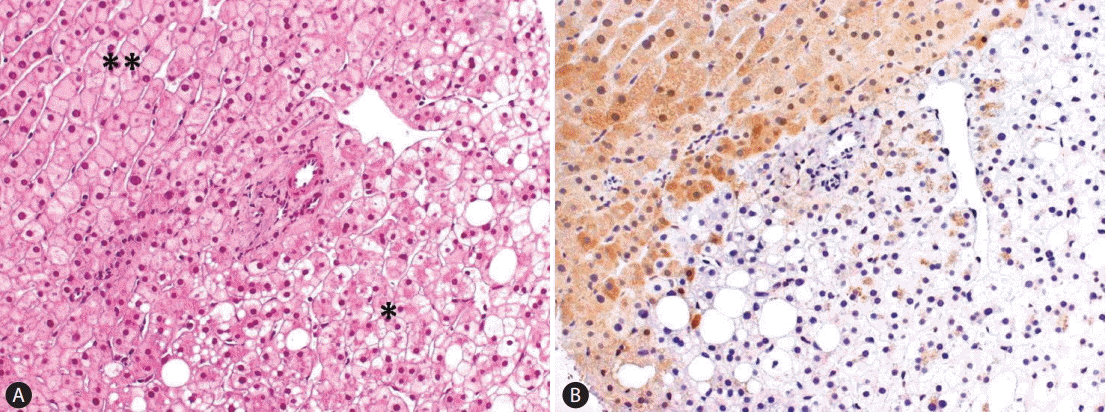
The first group of HCAs is defined by somatic inactivation of HNF1A gene, by a mutational mechanism in tumor cells, and accounts for 30% of HCA. HNF1A is a transcription factor controlling hepatocyte metabolism. Most H-HCA show macrovesicular steatosis of variable extent and no atypical hepatocytes and are associated to metabolic syndrome. A few tumors arise in patients carrying a germline HNF1A mutation in one allele which is associated with maturity-onset type 3 diabetes (MODY3). In this setting, adenomas undergo a second somatic mutation inactivating the second allele in the tumoral hepatocytes. Patients with germline mutations of HNF1A are predisposed to develop liver adenomatosis. Expression of liver fatty acid-binding protein (LFABP) involved in lipid trafficking, usually expressed in the normal liver, is specifically downregulated in H-HCA (100% accuracy) as a consequence of HNF1A mutation and serves as a translational marker to identify H-HCA (Fig. 2).
Inflammatory HCA (I-HCA)
The second group of HCA corresponds to inflammatory adenomas and accounts for 40% to 50% of all HCAs. Inflammatory syndrome, obesity and alcohol assumption are reported in these patients. These HCAs show the greater morphological polymorphism as they may show pseudo-portal tracts (fibrous tissue containing muscular-coated vascular structures and bordered by ductular reaction), sinusoidal dilatation, dystrophic arteries, hemorrhage, and inflammatory infiltrates. Steatosis is unusual but it may occur. The cardinal feature of these tumors is the activation of the JAK/STAT pathway. They also exhibit overexpression of Serum Amyloid Alpha (SAA) and C-Reactive Protein (CRP), two proteins of the acute phase of inflammation driven by inflammatory cytokines and chemokines, induced by STAT3. Five different molecular drivers, namely IL6 signal transducer (coding for gp130, mutated in 60% of I-HCA), FRK (10%), STAT3 (5%), GNAS (5%), JAK1 (1%) [5,41] have been reported. Each mutation is exclusive from the others and the 5 mutated genes are involved in more than 80% of the overall I-HCAs. CRP and SAA are the translational markers used to identify this particular type of adenoma (Fig. 3). CRP immunoreactive nodules in cirrhosis have also been detected and claimed to be a sort of I-HCA associated to alcohol assumption[42]. Notably, cases of inflammatory adenoma can also show activation of the ╬▓-catenin pathway (by gene mutation), which characterizes the third group of adenoma and identifies a subset of tumors more prone to undergo a malignant transformation. In these cases, atypical hepatocytes and features of atypical adenoma can also be seen.
╬▓-Catenin Mutated HCA (╬▓-HCA)
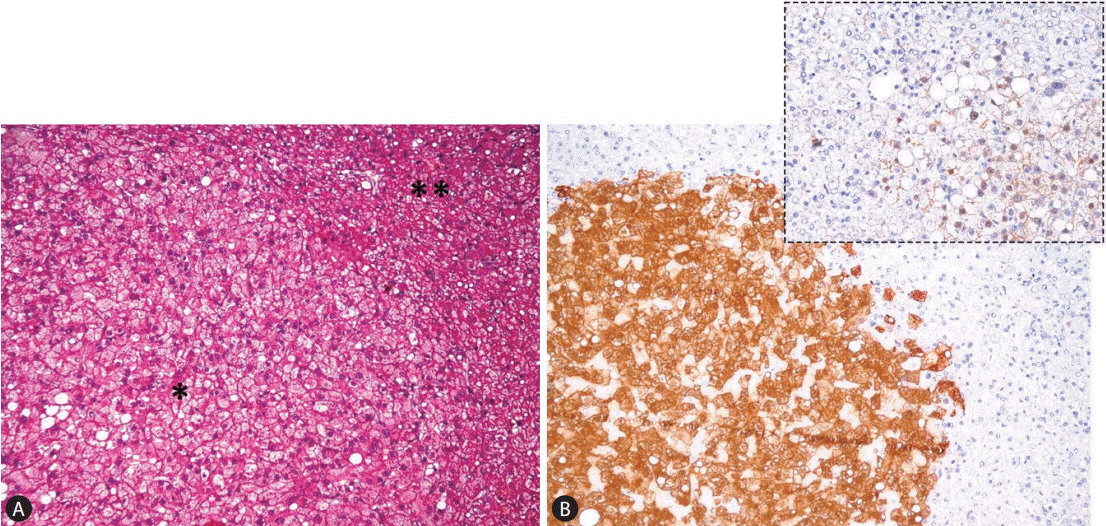
The third group is the ╬▓-catenin-mutated HCA (╬▓-HCA), which constitute approximately 10% to 15% of all HCAs. Not only females but also males (in some cases affected by congenital metabolic disturbances -glycogenosis- or assuming anabolic steroid), can develop ╬▓-HCA. Morphologically these tumors have cytological and architectural atypical features of tumoral hepatocytes, and cholestasis as well. The extent of atypical features, however, is limited and insufficient for the diagnosis of HCC. Mutations in the CTNNB1 gene coding for ╬▓-catenin are localized at hot spots in exon 3. Interestingly, ╬▓-catenin mutations are exclusive of HNF1A mutations, but half of ╬▓-HCAs are also inflammatory (╬▓-IHCA) and mostly characterized by concomitant gp130 or GNAS mutations. Importantly, ╬▓-catenin mutations are associated with a high risk of malignant transformation with HCC developing from this type of adenoma or elsewhere in the liver. ╬▓-HCAs show strong overexpression of GLUL (coding for GS), a target gene as revealed by quantitative reverse-transcription polymerase chain reaction analysis. The diagnosis of a b-HCA is made using bcatenin and GS immuostaining as translational markers. ╬▓-HCAs are characterized by a strong, homogeneous, non-map-like cytoplasmic expression of GS and, at the best, a nuclear and cytoplasmic immunohistochemical staining of ╬▓-catenin (Fig. 4). Other hot spot ╬▓-catenin mutations at exons of 7-8 have also been reported [41]. Immunohistochemical analyses of such cases revealed a faint and patchy GS expression without nuclear staining of ╬▓-catenin similar to that often seen in high grade dysplastic nodules arising in liver cirrhosis. Likely CTNNB1 mutants at exons 7-8 weakly activate ╬▓-catenin in vivo in HCA. As translational markers of ╬▓-catenin mutations, ╬▓-catenin nuclear staining and GS overexpression have absolute specificity but still suboptimal sensitivity (75% to 85%).
Unclassified HCA (u-HCA)
Unclassified adenomas, the last group of HCA represents approximately 10% of all HCAs. By definition, they lack the characteristics of the other subtypes and their identification relies on a silent phenotype and by the exclusion of criteria featuring the other subtypes. Until now, their underlying pathogenesis remains unidentified.
This molecular classification is used by pathologists in routine practice to classify HCA on resected specimens as well as on needle biopsies [20,34,43]. The translational markers such as LFABP, ╬▓-catenin, GS, SAA and CRP have been adopted as a panel to support a diagnostic conclusion of adenoma subtype and distinction from FNH. The interpretation of immunohistochemistry is usually carried out by comparing results in the tumor vs. those in the non-tumoral tissue. It is worth mentioning that LFABP should be absent in the tumor and present in the non-tumoral liver to specifically characterize the studied HCA as H-HCA. SAA and/or CRP should be diffusely positive in the tumor and largely absent in the non-tumoral liver to identify the HCA as IHCA. Finally, for ╬▓-HCA or ╬▓-IHCA, GS must be diffusely positive and not maplike, also in agreement with the presence of ╬▓-catenin nuclear staining. In the absence of clear-cut immunostaining, molecular biology as a gold standard is always necessary.
Diagnosis
HCA lesions are mainly discovered following abdominal pain and incidentally on imaging-based exploration. Sometimes, complications such as hemorrhage or malignant transformation can also reveal HCAs [38,44,45]. Liver enzyme levels are often normal, although anicteric cholestasis and inflammatory syndrome could be seen in association with I-HCAs [16]. Routinely tested tumoral markers are normal. Histologic analysis is the milestone for the diagnosis of HCA, but diagnosis between HCA and FNH or HCA and well-differentiated HCC can still be challenging, even for a dedicated liver pathologist. Histologic analysis of resected or biopsied hepatocellular tumors developed in normal liver is refined and supported by the use of a panel of markers qualified through the HCA molecular classification (GS, ╬▓-catenin, LFABP, PCR, and SAA) [20,34,46]. Intralesional morphology supplemented by special stains (reticulin, vascular profile, immunomarkers panel) can help to distinguish HCA from HCC in the majority of cases, as it will be discussed later. Despite this, unusual well-differentiated lesions can pose significant diagnostic issues even among liver pathologists. In these cases, the use of immunohistochemical biomarkers of malignancy such as GPC3, GS and HSP70 has been proposed as a useful tool to support the diagnosis [46,47]. For entities with definitely equivocal pathologic features the term of well-differentiated hepatocellular neoplasm of uncertain malignant potential (HUMP) instead of atypical hepatocellular neoplasm or atypical adenoma has been recently proposed [48]. However, the introduction of an undetermined borderline entity can have the side effect to formalize a basket where to put clinically heterogeneous entities of variable challenge in the diagnostic interpretation [49]. If confirmed the identification of TERT (telomerase reverse transcriptase) promoter mutation can be of help in these cases, as this event has recently been demonstrated to be a necessary second step to the full expression of histologic criteria of malignancy in ╬▓-catenin mutated adenoma [41,50].
Concept of atypical adenoma
Although rarely (<10%), HCA can transform into HCC or contain malignant foci or is associated with HCC [38]. Some clinical, pathological and molecular conditions are risk factors for malignant transformation, and are used to feature HCA as atypical. In the daily practice, the presence of at least one of these criteria is sufficient for a diagnosis of atypical adenoma and accordingly, it should be reported for the proper management, as illustrated in Table 1. We emphasize that the pathological features of atypical adenoma at routine staining have to be insufficient for a conclusive diagnosis of HCC. Interestingly, the use of a panel of 2 immunomarkers (HSP70 and GS) has been shown very useful to separate atypical (60% of the cases showing at least 1 of the markers) from typical adenoma (0%) [46]. The combined expression of both of these markers seems also useful in the diagnosis of very well-differentiated HCC, as their expression is more often increased in this category, as compared to atypical adenoma (38 vs. 10%) [46]. Finally, TERT promoter mutations have been reported in HCC arisen in HCA, but never in classical and atypical HCA [51].
Radiology also plays a prominent role, with specific techniques allowing non-invasive diagnosis in a large number of cases. On US, CT and MRI the key-imaging feature of HNF-1A inactivated HCAs is the presence of marked and diffuse fat within the lesion, respectively visible as homogeneous hyperechoic lesions (US) strong hypo-attenuation (unenhanced CT) and diffuse and homogeneous signal dropout on opposed-phaseT1-weighted sequences (MRI) [36,37]. The key-imaging feature of I-HCA is the presence of telangiectatic components, hypoattenuating and often heterogeneous at CT, hyperintense on T2-weighted and strong iso-to hyperintense on T1-weighted images at MRI [52].
Prognosis and treatment
Two main complications can affect HCA: hemorrhage and malignant transformation in HCC [53-55]. Even if rare, these events are difficult to predict and potentially life-threatening. The risk of hemorrhage is directly correlated with the size of the tumor, and Ōēź5 cm HCAs have a high risk of hemorrhage [38,53]. The risk of malignant transformation varies between 4% and 8% in the largest series [31,54,56]. However, most of the series that report malignant transformation focused on HCA-treated resection [54]. This could lead to an overestimation of the risk of malignant transformation [55]. In all cases, oral contraception or intake of androgen should be discontinued at diagnosis of HCA because regression of HCA has been described particularly after withdrawal of hormones. In HCA that do not regress after the withdrawal of oral contraception or androgen, surgical treatment has been the gold standard for a long time [57-59].
However, widely accepted guidelines for the management of patients with HCA are still lacking. In the past 10 years, a more conservative approach has been proposed by several centers to selectively adjust treatment to patients stratified by the risk of complications [38,44,60-62]. The diagnostic uncertainty with a well-differentiated HCC is an indication for surgical resection. Surgical approach should also be modulated by the risk of hemorrhage or malignant transformation together with the molecular subgroup of HCAs assessed by using MRI and/or biopsy [33,38,54]. Because HHCAs showed the lower risk of malignant transformation, typical features of small H-HCAs at MRI in young women could avoid biopsy and lead to radiologic follow-up [36,63]. It should be emphasized that HCC can arise also in these lesion, as recently reported [64]. In the absence of typical features of H-HCA at MRI, a tumor biopsy should be proposed to search for ╬▓-catenin activation/mutation and to better assess the risk of malignant transformation [33,36,43]. Liver transplantation should be avoided except for patients with glycogenosis 1a-associated adenomatosis, where liver transplantation can manage both the HCA and the underlying metabolic disorder [65]. For adenomatosis, even despite the lack of robust clinical studies and consensus, the same rules as for sporadic HCAs should be applied considering for resections larger and ╬▓-catenin mutated lesions [38,66]. Even if bleeding from HCAs were sometimes observed during pregnancy, larger series have shown that pregnancy in patients with residuals, not at risk HCAs, is usually safe [38,67,68]. Thus, pregnancy should not be contraindicated but closely follow-up by imaging. In case of bleeding with hemodynamic instability, urgent arterial embolization could be proposed as a first-line treatment to block the hemorrhage [69]. Reduction in tumor size is frequently observed after arterial embolization, and this could also limit the extent of the resection. In a second step, surgical resection should be performed a few months later after embolization [30,70].
The diagnosis of FNH and HCA in the clinical practice
In the clinical practice, the diagnostic approach to these lesions is always clinical and radiological. HCA rather than FNH usually arises in a specific context (young female, use of oral contraceptive, metabolic disease, inflammatory syndrome, alcohol assumption etc.) which needs an extremely careful preliminary investigation. Sporadic adenomas unrelated to certain clinical traits/diseases are rare. Imaging also play a very important role particularly in ascertaining FNH where up to 80% of cases are usually and confidently diagnosed without additional diagnostic procedures. HCA is also increasingly characterized by imaging with special regards to certain types (H-HCA). Overall, given the greater heterogeneity of HCA as compared to FNH, radiologically doubtful FNH and the majority of HCA require the liver biopsy. Identification of FNH and HCA on surgical specimens is rare but it can also occur as a consequence of a non-conclusive histology in the liver biopsy, of an incidental finding in hepatic resections for primary or metastatic unrelated tumors or for the removal of an inaccurately characterized hepatic lesion from a clinico-radiological profile.
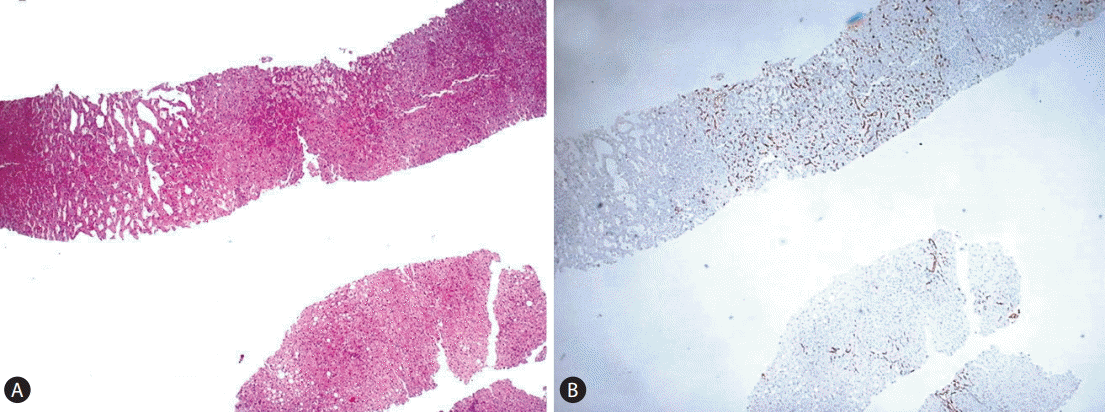
We propose the following diagnostic algorithm for the study of these lesions in the liver biopsy (Fig. 5). When dealing with a liver biopsy of a hepatocellular nodule in the healthy liver, the first question is whether the lesion under study has been adequately sampled, as to permit a diagnostic conclusion and not a mere (non-conclusive) morphological description. Although the size of the liver biopsy is important, the critical feature, regardless the biopsy size and thickness, is to have an available representative intralesional material of the hepatocellular component of the nodule. To address this issue, the availability of an extralesional or perilesional liver tissue is of great value because both FNH and HCA usually arise in the context of a normal healthy liver, without significant fibrosis or histological features of portal hypertension. On routine H&E stain both FNH and HCA may have a subtle and deceptive morphology as to that their outlines and peripheral borders may be difficult to be clearly localized. A good tool to highlight them, particularly in HCA, is to stain a section for an endothelial cell marker (we use CD34) which will permit in the majority of the cases to discern the profile of the punched lesion (Fig. 6). The increased vascular network supporting these lesions is in our hands a very good tool to assess sampling adequacy. A CD34-negative stain is against a good sampling of a FNH/HCA.
Once the lesion has been shown in the fragment(s), we usually evaluate whether pseudo-portal tracts (fibrous tissue with arteriolar vascular structures and ductular reaction), can be documented within the lesion. Pseudo-portal tracts should not be confused with true portal tracts (containing normal branch of hepatic artery, portal vein and interlobular bile ducts, without a ductular reaction) which may be trapped within the lesion at the borders. The presence of pseudo-portal tracts suggests two diagnostic alternatives: a) FNH, b) I-HCA. In the liver biopsy, a clear-cut distinction between these two entities is not feasible on pure H&E morphological grounds. However, FNH and I-HCA may be distinguished in most of the cases using a panel of 3 markers (GS, CRP and SAA). Accordingly, we can discern two alternative profiles: a) map-like GS+/SAA-/CRP- which militates against HCA, and supporting FNH and b) GS-/SAA+/CRP+ or non-map-like GS+/SAA+/PCR+ which militates against FNH, supporting I-HCA. In fact, GS immunoreactivity can be seen in those I-HCA also characterized by an activation of ╬▓-catenin pathway and in these cases, GS staining will not be map-like but strong and diffuse. In the cases also characterized by ╬▓-catenin gene mutation, nuclear ╬▓-catenin immunoreactivity will also show up at least in a few hepatocytes. These cases can also show the morphology of atypical adenoma where it will be necessary to evaluate the possible transition/transformation of the adenoma into a HCC.
When pseudo-portal tracts are not detectable inside the lesion and GS staining is completely negative, the alternative is between H-HCA and a u-HCA. H&E features will be of great help to guide the diagnostic algorithm. The evidence of macrovesicular steatosis in particular and/or of glycogenated nuclei will call for a case of H-HCA in which the diagnostic evidence will be given by the lack of expression of LFABP in tumoral hepatocytes. Results can be clearly documented in the liver biopsy particularly when extralesional LFABP+ hepatocytes are seen for comparison. Please note that a few LFABP+ non tumoral but entrapped hepatocytes can also be observed at the lesional borders. Conversely, the case of a non steatotic adenoma characterized by the absence of hepatocellular atypia and by a completely silent phenotype (LFABP-/SAA-/CRP-/GS-) will permit to classify the adenoma as u-HCA, a variant where a driver molecular alteration is still unknown. Finally, when no pseudo-portal tracts are detected inside the lesion while atypical hepatocytes, isolated or in groups, are clearly seen in the absence of an inflammatory/telangiectatic morphology and phenotype (SAA-/CRP-), the nodule is likely to be a ╬▓-HCA variant with GS and ╬▓-catenin staining positive. The former antigen, when the staining is strong and diffuse, shows the highest diagnostic accuracy (absolute specificity and sensitivity).


 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print




