


Hepatocellular carcinoma (HCC) is the most common liver cancer and the fourth most common cause of cancer mortality worldwide. Persistent infections with the hepatitis B and hepatitis C viruses have accounted for the etiology of majority of the HCC cases. Owing to the recent development of antiviral agents, the proportion of HCCs caused by viral hepatitis is decreasing. Direct-acting antiviral agents for hepatitis C dramatically improve the sustained viral response rate in patients with chronic cases, leading to a decrease in liver disease-related mortality in patients with chronic hepatitis C. Although antiviral treatment is effective for the prevention of virus-related HCC, the etiology of the cancer is shifting from viral hepatitis to lifestyle-related metabolic diseases, such as non-alcoholic steatohepatitis (NASH) [1].
Currently, surveillance for HCC has been conducted mainly for patients carrying the hepatitis virus, because the vast majority of HCC patients are positive for hepatitis B or C. In light of the trend of increasing metabolic disease-related HCCs, an effective program focusing on these diseases is required, which may be difficult to establish because of the high prevalence and variety of metabolic disorders, including impaired glucose tolerance, diabetes mellitus, and obesity [1,2]. In addition, metabolic diseases likely accelerate the emergence of HCC even after the elimination of the hepatitis virus. Nahon et al. [3] reported that the cumulative incidence of HCC development among patients with chronic hepatitis C who achieved the sustained viral response was stratified by the presence or absence of metabolic features defined by a body mass index (BMI) of >25 kg/m2 and/or diabetes mellitus and/or dyslipidemia. The population of both adults and children who are either overweight or obese continues to grow in high-income as well as low- and middle-income countries, indicating a further increase in metabolic disease-related features.
To clarify the effect of obesity on hepatocarcinogenesis, Sohn et al. [4] performed meta-analyses and studied the relationship between BMI and the emergence or mortality rate of liver cancer using data exclusively from a prospective cohort study, where the majority were likely to be HCC. They clarified the dosage effect of BMI on liver cancer emergence, where an increase in BMI was positively associated with the development of tumors, although the analysis was performed using the categorical variables for BMI [4]. In addition, they showed that higher BMI is related to the increase of HCC emergence and mortality even in subjects with hepatitis B or C. Although the cause-and-effect relationship between obesity and carcinogenesis is difficult to be demonstrated in human subjects, the analysis using a large number of cases from prospective cohort studies and the confirmation of the dosage effect of BMI on HCC emergence further supports the idea that obesity needs to be managed in the prevention of metabolic disease-related HCC and thus mortality. From this perspective, it is necessary to understand the pathogenesis of HCC caused by metabolic perturbations.
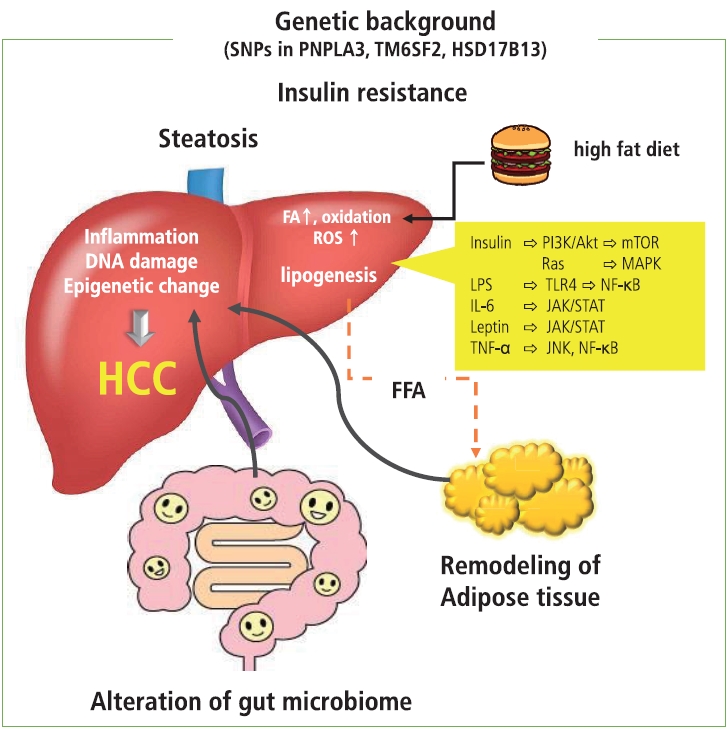
It is likely that several conditions, such as insulin resistance, hyperinsulinemia, remodeling of adipose tissue, alteration in intestinal flora, and an increase in endoplasmic reticulum and oxidative stress, act in concert to contribute to the development of metabolic disease-related HCC (Fig. 1).
Adipocyte inflammation occurs in obese patients with insulin resistance and hyperinsulinemia [5]. Under these conditions, secretion of adipokines from adipocytes that facilitate insulin sensitivity, such as adiponectin, is impaired. Generally, normal adipocytes secrete adiponectin, which promotes insulin sensitivity and fatty acid oxidation and suppresses lipogenesis. In obesity, adipocytes show swelling and dedifferentiation that lead to the impaired secretion of adiponectin, and subsequent infiltration of macrophages contributes to sustained inflammation. The excess amount of fatty acids induced by a high-fat diet results in the overload of oxidation in mitochondria, resulting in the generation of reactive oxygen species that promote DNA damage. Additionally, a high fat level induces inflammation in adipose tissue and liver through the increase of oxidative stress and activation of the transcription factors involved in the inflammation process, such as nuclear factor kappa B (NF-╬║B). In addition, hypertrophic adipocytes induce ischemia in adipose tissue, where the accumulation of inflammatory cells and increase of inflammatory cytokines occur [6], and are prone to release proinflammatory cytokines and chemokines, including tumor necrosis factor-╬▒, interleukin-6, C-C motif chemokine ligand 2, and resistin. These cytokines also accelerate insulin resistance through the activation of the Jun amino terminal kinase and NF-╬║B signaling; insulin resistance induced by proinflammatory cytokines, adhesion molecules, and transcription factors contribute to the development of the hepatic manifestation of the metabolic disease NASH [5,7,8].
On the other hand, obesity-induced inflammation as well as a high-fat diet accelerate the change in gut microbiomes and increase permeability of gut mucosa, leading to the leakage of inflammatory factors, including bacterial metabolites and other bacterial components showing microbiome-associated molecular patterns. These factors cause liver inflammation, injury, and DNA damage and eventually induce the progression of NASH and the development of metabolic disease-related HCC. Indeed, some reports show that microbial transfer from obese mice could induce the progression of NASH in the lean mouse model [9,10]. Furthermore, it has been reported that an increase in alcohol-producing bacteria in the intestinal flora may cause liver injury. In addition, alteration of the gut microbiome results in the gut flora producing secondary bile acid through the function of bacterial bile acid 7╬▒-dehydroxylase. Secondary bile acids, deoxycholic acid, and liothocholic acid are known to induce carcinogenesis through the induction of inflammation and oxidative stress. These types of secondary bile acids could affect hepatic satellite cells and induce a senescence-associated secretary phenotype via the stimulation of the toll-like receptor 2 [11,12]. The phenotype alteration in the hepatic satellite cells induces the production of prostaglandin E2 and suppresses antitumor immunity [12]. A high-fat diet impairs the function of the farnesiod X receptor that plays a role in hepatic triglyceride homeostasis, glucose stage and release, and acceleration of the induction of steatosis [13]. These conditions cooperate and contribute to the development and progression of inflammation, fibrosis, and hepatocarcinogenesis through the alteration of cellular signaling, such as the activation of phosphoinositide 3-kinase (PI3K)/Akt, NF-╬║B/mammalian target of rapamycin complex (mTOR), and Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathways in hepatocytes as well as mesenchymal cells.
In addition to adipocyte inflammation, insulin resistance, and changes in the gut microbiome, several genetic and epigenetic backgrounds have been reported in association with NASH and metabolic disease-related HCC. A genome-wide association study revealed that the single nucleotide polymorphism (SNP) rs738409 C >G in the patatin-like phospholipase domain-containing 3 gene, which plays a role in the regulation of lipolysis in hepatocytes, is associated with the increased risk of developing NASH and HCC [14]. The SNPs in the transmembrane 6 superfamily member 2 gene and hydroxysteroid 17-beta dehydrogenase are also known to be associated with metabolic disease-related HCC [15,16]. The combination of SNPs in the phospholipase domain-containing 3, transmembrane 6 superfamily member 2, and hydroxysteroid 17-beta dehydrogenase genes reportedly predicts a 29-fold increased risk for HCC, suggesting that genetic factors have a fundamental role in the development of metabolic disease-related HCCs [17]. Moreover, metabolic perturbation has been reported to result in the alteration of epigenetic modification in a mouse model. For example, glycine N-methyltransferase knockout mice, which develop NASH and HCC, show hypermethylation on the promoter of the tumor suppressor Ras-association domain family member 1, suppression of the cytokine signaling 2 gene, and activation of the Ras and JAK/STAT signaling pathway [18]. Additionally, hyperinsulinemia may affect histone modification and contribute to the development of HCC through the altered expression of genes involved in cellular signaling [19]. These epigenetic alterations can lead to the dysregulation of adipokine secretion and activation of PI3K/Akt, JAK/STAT, NF-╬║B, mTOR, trans-4-hydroxy-2-nonenal, and the nuclear factor erythroid 2-related factor 1 oncogenic pathway in hepatocytes. Alteration of DNA methylation and histone modification induces the activation of noncoding RNAs such as miR-21, miR-221, and miR-222, which are responsible for cell proliferation and carcinogenesis [20].
In summary, obesity is a major cause of inflammation in adipose tissue as well as the liver; additionally, it can induce insulin resistance, as well as affect the immune systems in the gut and liver. Some reports show immunological aspects of metabolic disease-related HCC and its treatment [21,22]. These conditions affect the gut microbiome, modulate the histone code, and alter the expression of noncoding RNA. Certain genetic conditions can accelerate the progression of obesity-induced disorders. As metabolic perturbations affect gene expression and molecular alteration, and vice versa, making efforts to understand the influence of obesity and lifestyle events is becoming critical for achieving effective management of the emergence of metabolic disease-related HCC.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print





