


INTRODUCTION
Hepatocellular carcinoma (HCC) is the second leading cause of cancer-related deaths globally. HCC, the primary liver cancer, is regarded as the third most common malignancy worldwide [1,2]. The major risk factors of HCC includes hepatitis C and B viral infections, fungal metabolite aflatoxin A1 exposure, alcohol intake, obesity, and non-alcoholic fatty liver diseases [3,4]. HCC is generally asymptomatic in nature and hence, is mainly diagnosed in the advanced stage [5]. The lack of primary screening leads to an increased mortality to incidence ratio. Furthermore, sorafenib, an orally active multikinase inhibitor, is the first-line of therapy showing a significant survival benefit in patients with advanced HCC. Sorafenib has a direct effect on the components of tyrosine kinase cell signalling pathways which are generally deregulated in HCC [6,7]. Sorafenib remarkably prolonged the survival rates in advanced HCC patients as illustrated by randomized controlled clinical trials. However, the management of HCC is still debatable [8]. The reason behind this, is the development of adaptive drug resistance against sorafenib therapy [9,10]. Global analysis of the expressed cellular proteins may aid to identify the differentially expressed proteins between parental cells and drug resistant cancer cells. Besides, malignant cell culture models are also suitable for application of proteomic techniques in order to recognize particular protein that might be associated with a characterized phenotype of the malignant cells [11].
Recent studies have demonstrated that various alterations in the cytoskeletal proteins may be one of the important mechanisms involved in the development of drug resistance in cancer cells that may in turn be associated with altered drug efflux pumps [12]. Among them, vimentin is the cytoskeletal protein which belongs to the family of intermediate filament and is one of the markers of epithelial-mesenchymal transition (EMT) [13]. The vimentin expression in cancers is associated with increased tumor invasion and proliferation. Deregulation of vimentin expression and its relation to tumor metastasis has been discussed in various cancers such as gastrointestinal tumors [14,15], prostate carcinoma [16,17], and breast cancer [18,19]. A relation between vimentin expression and drug resistance has been demonstrated in ovarian cancer as the downregulation of vimentin is associated with acquired drug resistance to cisplatin [20]. Sorafenib-resistant cancer cells may undergo EMT, however, various studies have shown that sorafenib downregulate this process in HCC cells. During the exposure of mouse primary hepatocytes to sorafenib, EMT gets diminished due to decrease in transforming growth factor ╬▓ signalling [21]. A study by van Malenstein et al. [10] in 2013, also have shown that long term exposure of sorafenib to HepG2 cells leads to development of resistance due to activation of EMT. But no such study has yet been revealed EMT cytoskeleton proteins as a potential target for treatment along with sorafenib. Further, the exact role of vimentin is not well established in sorafenib-resistant HCC.
In this study, we have unveiled the expression pattern of vimentin in parental as well as sorafenib-resistant HepG2 cells and further, highlighted the significance of vimentin as a plausible therapeutic target in sorafenib resistant HCC model cell line, HepG2.
MATERIALS AND METHODS
Cell lines and cell culture
This study was performed on HepG2 cell line that procured from cell repository of National Centre for Cell Sciences, Pune, India. HepG2 cells were then cultured in minimum essential media (MEM) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS), 0.15% (v/v) sodium bicarbonate, 1 mM sodium pyruvate and 2 mM L-glutamine. HepG2 cells were maintained at 37ºC in 5% CO2 and 95% humidity in a CO2 incubator (Thermo Heraeus HERAcell® 240; Thermo Scientific, Waltham, MA, USA). Antibiotics streptomycin (100 μg/mL) and penicillin (100 U/mL) were supplemented in the media for maintenance of the cultures.
Chemicals
The media and antibiotics for cell culture were procured from HiMedia (Chandigarh, India), Sigma-Aldrich (St. Louis, MO, USA), and ThermoFisher Scientific. Sorafenib was procured from Santacruz Biotechnology, Inc. (Dallas, TX, USA). Withaferin A was procured from Cayman Chemicals Co-USA (Cayman Chemical, Ann Arbor, MI, USA). Both sorafenib and withaferin A were dissolved in dimethyl sulfoxide (DMSO) to prepare 1 mM stock and 20 mM stocks respectively for further use in cell lines.
Samples preparation for matrix-assisted laser desorption/ionization (MALDI)
The total protein isolated from both HepG2 parental [HepG2 (P)] and HepG2 sorafenib-resistant [HepG2 (R)] cells was estimated following bicinchonic acid method (Sigma-Aldrich). The proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) according to the method of Laemmeli (1970). After washing the gel with water, the spot of interest was excised and 200 ┬ĄL of distaining solution was added to the gel, vortexed and incubated for 5 minutes and then the supernatant was discarded in order to destain the gel. Gel particles were then treated with 10 mM dithiotreitol/50 mM NH4HCO3 (freshly prepared), followed by incubation for 45 minutes at 56┬║C and then immediate cooling of tubes at room temperature. The gel pieces were treated with light sensitive 55 mM idoacetamide prepared in freshly prepared 50 mM NH4HCO3 for 30 minutes at room temperature and then washed with wash buffer. Freshly prepared trypsin enzyme solution (30 ng) was added to the gel, incubated at 37┬║C overnight. Following overnight incubation, the supernatant was collected and 5 ╬╝L of extraction buffer containing 60% ACN+ 0.1% TFA in 25 mM NH4HCO3 was added to the gel, vortexed and incubated for 10 minutes. Supernatant was collected into the same tube in which supernatant collected from trypsin digestion was added. The above process was then repeated first with extraction buffer containing 70% ACN+ 0.1 TFA in 25 mM NH4HCO3 and then with 90% ACN+ 0.1% TFA in 25 mM NH4HCO3. The extracted peptides were stored at ŌĆō20┬║C for MALDI analysis.
MALDI-time of flight (TOF) mass spectroscopy
MALDI spectra were acquired using an Ultraflex TOF/TOF mass spectrometer (BrukerDaltonic, Hamburg, Germany). The resulting peaks were subjected to MASCOT search (www.matrixscience.com) and parameters were set as carbamidomethyl modification of cysteine, one missed cleavage was allowed for trypsin (SigmaAldrich). Once protein was identified it was further fragmented and subjected to MS-MS analysis. Protein MASCOT score, sequence coverage, and number of peptides matched were used to confirm the protein using SwissProt database (www.uniprot.org).
RNA isolation, cDNA synthesis and quantitative real-time polymerase chain reaction (qRT PCR) analysis
Total RNA was isolated from both HepG2 (P) as well as HepG2 (R) cells using the Trizol reagent (Invitrogen, Carlsbad, CA, USA). Finally, RNA concentration was quantified and processed for cDNA synthesis. cDNA synthesis was carried out from the intact RNA by using Verso cDNA Synthesis Kit (Thermofischer). qRT PCR was performed on Lightcycler┬« 96 (Roche, Bremen, Germany) using SYBR Green I master (Roche) detection method to check relative expression of target genes using specific primers for glyceraldehyde 3-phosphate dehydrogenase (GAPDH), vimentin and ABCG2 genes. The relative mRNA levels were calculated using the formula 2-ŌłåŌłåCt, where ŌłåCt = (Ct target gene - Ct internal control). Each set of primers were designed to target specifically exon-exon junction in cDNA and was commercially synthesized by Sigma-Aldrich, USA. The gene expression was normalized using GAPDH acting as internal control. The primer sequences are:
ABCG2 (F)-GTGGCCTTGGCTTGTATGAT (R)-GATGGCAAGGGAACAGAAAA
GAPDH (F)-CCATCTTCCAGGAGCGAGA (R)-GGTCATGAGTCCTTCCACGAT
Vimentin (F)-CCGGTGCAATCGTGATCTCTGGG (R)-ATTCAAGTCTCAGCGGGCTC
Protein expression: flow cytometry
HepG2 (P) and HepG2 (R) cells were cultured in a 12-well plate. After 24 hours of treatment, cells were trypsinized and washed twice with phosphate-buffered saline (PBS) in dark and cell pellet was resuspended in PBS. The cells were then fixed using 4% paraformaldehyde solution and again washed using PBS. After washing, the cells were permeabilized using permeabilizing buffer (1% BSA + 0.01% Triton X 100 + 0.01% Sodium azide in 1X PBS) and washed with PBS. Then blocking buffer (5% BSA in 1X PBS) was used to blocking the cells. Antibody solution was added after blocking to cells and incubated for 1 hour at room temperature. The expression of vimentin was then analyzed using flow cytometry.
MTT assay
HepG2 (P) and HepG2 (R) cells were seeded in a 96-wells plate (Corning Inc, Corning, NY, USA) with density of 5,000 cells per well in MEM with 10% FBS. After 24 hours of incubation, various concentrations (conc.) of withaferin A and sorafenib were added to MEM with 0.5% FBS, a total volume of 200 ┬ĄL per well. After 24 hours, sterile 20 ┬ĄL MTT solution was added to each well and plates were incubated for 4 hours at 37┬║C, 5% CO2. Media containing MTT was then removed from the each well and formazan crystals formed were dissolved in DMSO (200 ┬ĄL) and incubated for 5 to 15 minutes at room temperature in dark. The absorbance was then measured at 570 nm using a microtiter plate reader by infiniteM200PRO microplate reader (Tecan, M├żnnedorf, Switzerland).
Crystal violet assay
This analysis was done to assess the growth inhibition pattern of HepG2 (P) and HepG2 (R) cells under different conditions. HepG2 cells (2,000 per well) were seeded in a 24-well plate in triplicate. After 24 hours of treatment, the cells were washed with PBS (1X) and later fixed using 4% paraformaldehyde in PBS (1X) for 15 minutes (200 ╬╝L per 12-well). Then, the cells were stained with 0.1% crystal violet (50 mg crystal violet powder in 5 mL ethanol/45 mL water) for 20 minutes (100 ╬╝L per 12-well). Subsequently, after washing with PBS, 500 ╬╝L 10% acetic acid was added to each well and incubated for 20 minutes on shaker. Finally, out 0.5ŌĆō1 mL of stain was taken out and absorbance was checked at 590 nm.
Statistical analysis
For the statistical analysis the experimental group differences were calculated by StudentŌĆÖs t-test or one-way ANOVA with BonferroniŌĆÖs correction for multiple comparisons. Drug concentrations effect in two cell types were assessed by two-way ANOVA with SidakŌĆÖs correction. All data were analyzed by GraphPad Prism (v.4.00; GraphPad Software, San Diego, CA, USA). Data were presented as the mean and standard error of the mean. Significance level was set at 0.05.
RESULTS
Stored HepG2 (P) and HepG2 (R) cells were recultured to perform the present experiments. As earlier (Ankita Makol, Ph.D. thesis, 2017) the HepG2 cell line was treated with minimal concentrations of sorafenib inhibitory concentration (IC)-20 dissolved in DMSO, continuously for long period followed by further fractional dose elevation to mimic the clinical settings. As a result, sorafenib-resistant HepG2 cell line was established [22]. The chemoresistance nature of HepG2 cells was characterized in view of microscopic analysis (Fig. 1A) and dose-response assessment (IC-50) in presence of different doses of the sorafenib (Fig. 1B). The IC-50 value of resistant cells was 2.25 fold higher than parental cells.
Protein profiling and identification of differentially expressed proteins in HepG2 cells
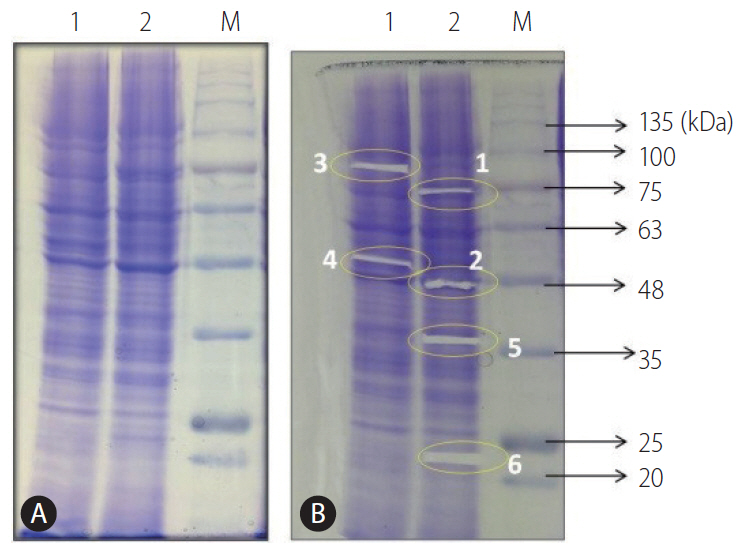
In order to detect candidate protein marker which may have role in development of resistance against sorafenib, the comparative protein profiling of HepG2 (P) and HepG2 (R) cells was done using SDS-PAGE followed by coomassie staining (Fig. 2A). The six fragments (1, 2, 3, 4, 5, and 6) on the coomassie stained gel (Fig. 2B) were selected for MALDI-TOF-MS analysis. Among them peptide mass fingerprinting (PMF) of each in-gel digested four samples along with the respective theoretical weight, protein coverage, peptide matches which were documented from Swiss-Prot for each samples (Supplementary Table 1). The identified proteins were further confirmed by MS analysis. The analysis resulted in the identification of four proteins which were differentially expressed in HepG2 (P) and HepG2 (R) cells.
According to PMF analysis, sample 1 (excised protein band) was identified to be glucose regulated protein 78 (GRP78) which is highly expressed in HepG2 (R) as compared to HepG2 (P) cells whereas sample 2 was identified to be actin, highly expressed in HepG2 (R) in comparison to HepG2 (P) cells. On the contrary, sample 3 was identified to be TRAP1 that was highly expressed in HepG2 (P) cells as compared to HepG2 (R) cells. Sample 4 was identified to be vimentin with theoretical mass 50 kDa which was similar to observed molecular weight (53,676 Da), protein coverage 44%, peptide matches 31. Vimentin was further confirmed by MS-MS analysis. Two peptides of molecular weight 1,572 Da and 1,094 Da were analysed which showed the identification with peptides of vimentin which was highly expressed in HepG2 (P) cells in comparison to HepG2 (R) cells (Supplementary Fig. 1).
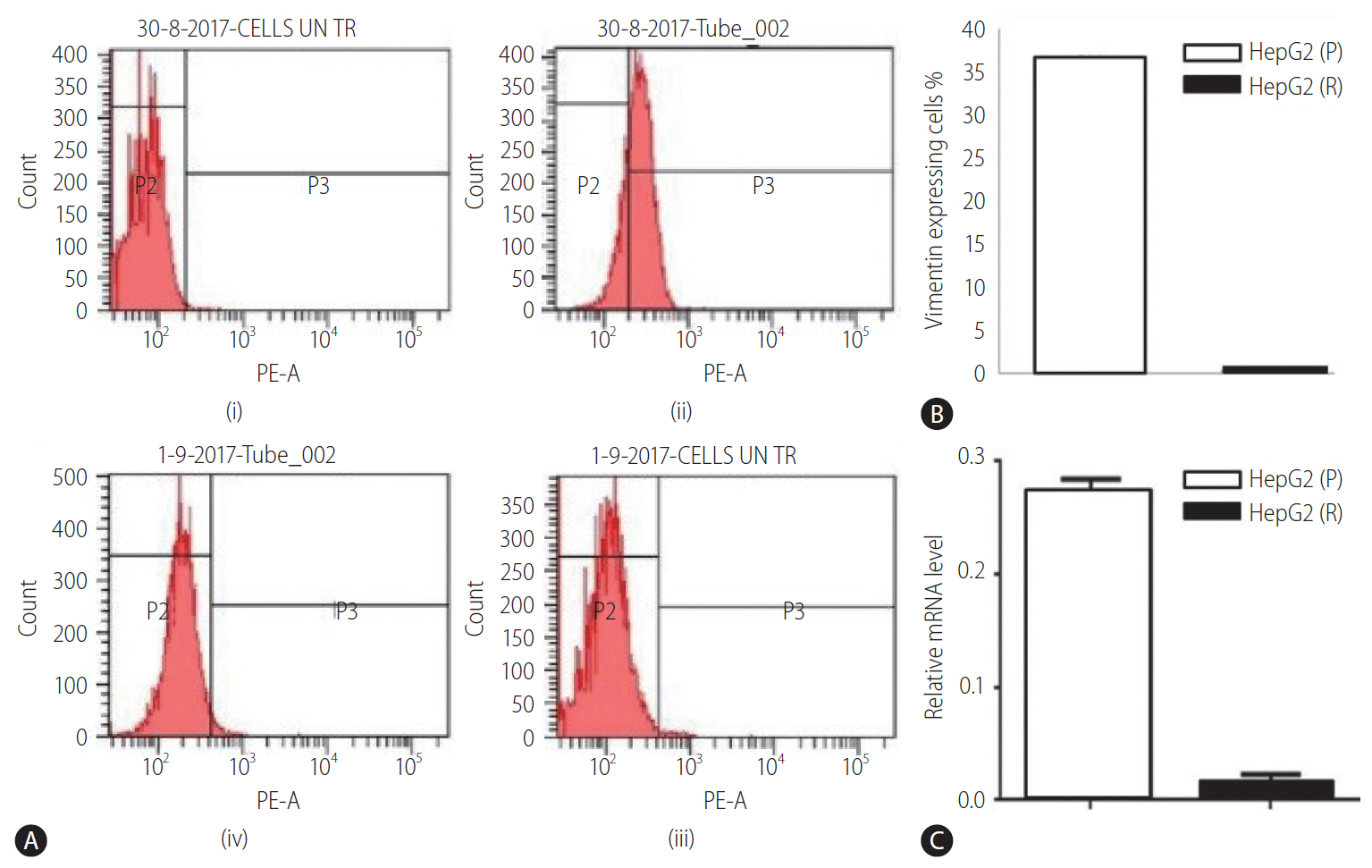
Expression of vimentin in HepG2 (P) and HepG2 (R) cells
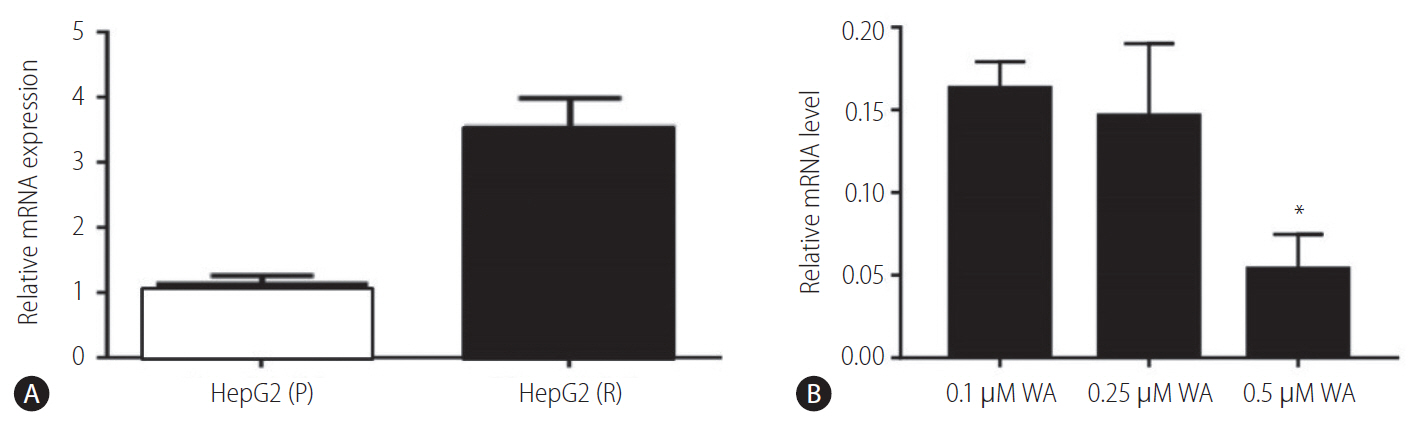
The expression level of vimentin was examined in both HepG2 (P) and HepG2 (R) cells at mRNA levels by qRT-PCR. Vimentin gene expression was resulted to be downregulated in HepG2 (R) cells in comparison to HepG2 (P) (Fig. 3C). These results were further confirmed at protein level through analysis of vimentin by flow cytometry. The protein expression analysis using phycoerytherin labelled anti-vimentin antibodies (mouse anti-human antibodies) revealed higher expression of vimentin in HepG2 (P) cells as compared to HepG2 (R) cells (Fig. 3A, B).
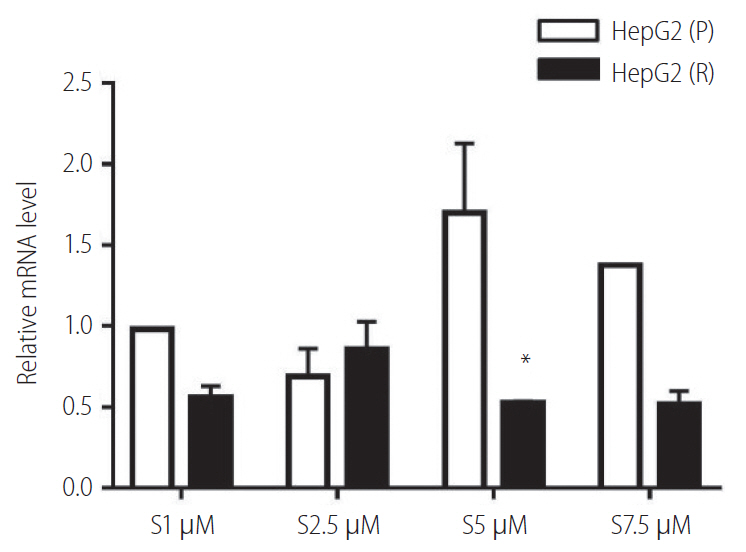
Vimentin expression in response to sorafenib
In the presence, of different concentrations of sorafenib (1.0, 2.5, 5.0, and 7.5 ┬ĄM), the relative expression levels of vimentin in HepG2 (P) and HepG2 (R) cells has shown deregulated behaviour. Moreover, at sorafenib conc. 5 ┬ĄM, vimentin expression was significantly decreased in HepG2 (R) cells. Therefore, this concentration was selected for further experiments (Fig. 4).
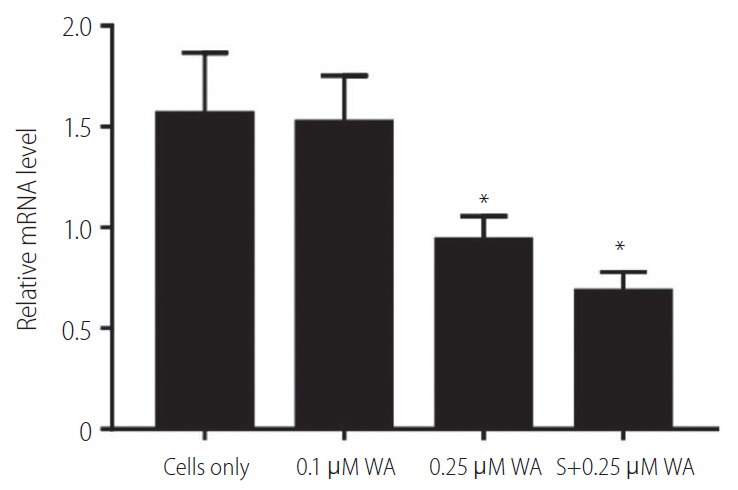
Vimentin expression in relation to withaferin A and its combination with sorafenib
Withaferin A is a steroidal lactone found in the restorative plant, Withania somnifera (Ashwagandha). It is reported as a potent inhibitor of vimentin, as it is known to cause the aggregation of tetrameric form of vimentin [23]. We found that gene expression levels of vimentin was downregulated in the HepG2 (R) cells when treated with increasing concentrations of withaferin A. In HepG2 (R) cells, the vimentin expression was significantly decreased at withaferin A drug concentration of 0.25 ┬ĄM (IC-50 value; >6 fold decrease).
Further, we also examined the gene expression of vimentin in HepG2 (R) cells in response to the combination of withaferin A and sorafenib as compared to the untreated ones. It was observed that when HepG2 (R) cells were treated with the combination of sorafenib (5 ┬ĄM) and withaferin A (0.25 ┬ĄM; IC-50 value), the gene expression of vimentin showed a significant decrease in comparison to untreated cells (Fig. 5).
Effect of vimentin inhibition on sorafenib resistance: expression analysis of ABCG2
ABCG2 transporters are known to be commonly associated with the development of drug resistance in majority of cancers [24]. Elucidating the targeted effect of withaferin A on the ABCG2 gene expression in HCC cells, we analyzed the effects of its incremented doses as well as in combination with sorafenib.
As expected, the ABCG2 gene expression was higher in HepG2 (R) cells as compared to HepG2 (P) cells (Fig. 6A). Further, the levels of ABCG2 gene were observed to decrease with 0.25 ┬ĄM (IC-50 value) withaferin A treatment in HepG2 (R) cells and at drug concentrations of 0.5 ┬ĄM, the levels of ABCG2 were observed to decrease significantly (Fig. 6B).
Effect of vimentin inhibition on cell viability of HepG2 cells
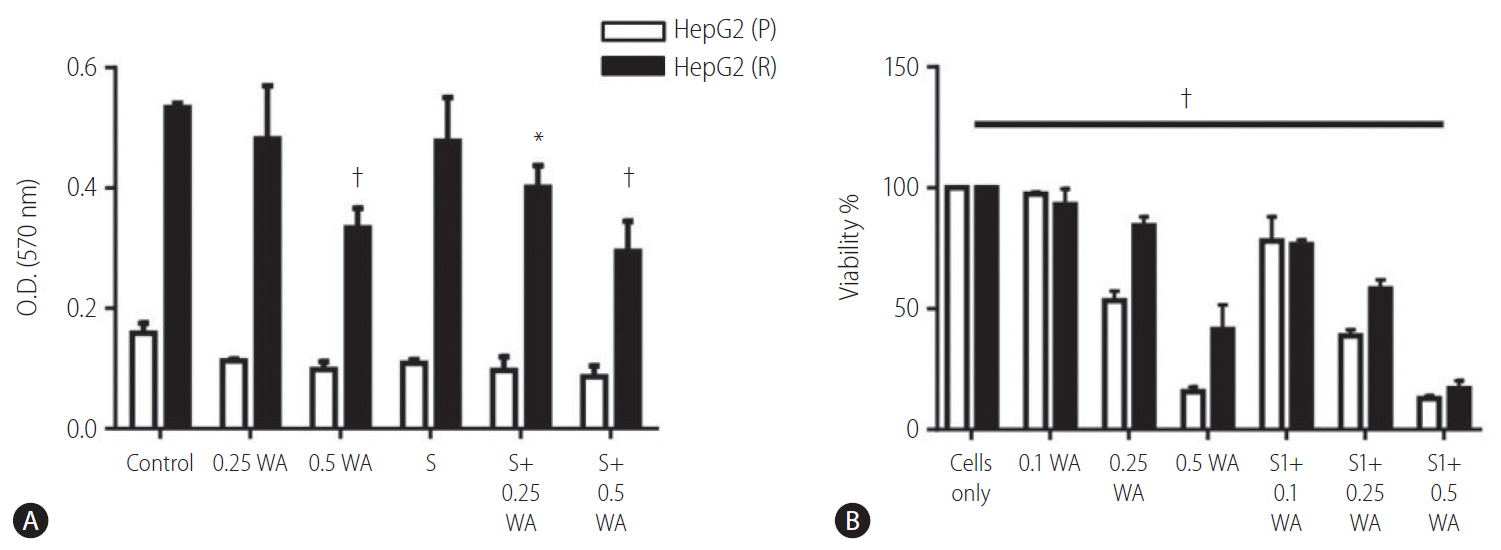
The effect on cell viability via withaferin A treatment and combination of withaferin A with sorafenib was assayed through crystal violet staining. This analysis depicted the dose dependent cytotoxic effect of withaferin A and combination of withaferin A with sorafenib. The cell number significantly decreased with increasing concentration of withaferin A as well as with the combination of sorafenib and withaferin A (Fig. 7A).
The cell viability of HepG2 (P) and HepG2 (R) cells was also confirmed through the MTT assay. On treating the cells with different concentrations of withaferin A as well as in combination with sorafenib (5 ┬ĄM), the cell viability was found to decrease significantly in both HepG2 (P) and HepG2 (R) cells (Fig. 7B).
As increasing concentration of withaferin A as well as its combination with sorafenib, the only first-line therapy for advanced HCC has a direct effect on cell viability of both HepG2 (P) and HepG2 (R) cells.
Hence, inhibition of vimentin might involve modulating sorafenib resistance as well as proliferation of HCC.
DISCUSSION
HCC is the primary cancer of liver and the leading cause of death in patients with liver diseases. The first-line systemic therapy of choice for advanced HCC is sorafenib that is known to extend the median survival time moderately by 2ŌĆō3 months. However, long-term exposure of sorafenib has been reported to induce adaptive resistance in patients. The main processes which are known to have a role in sorafenib resistance broadly include hypoxic microenvironment, autophagy and EMT etc. EMT is a process of acquiring more metastatic and invasive properties similar to mesenchymal cells and is stated as losing the normal epithelial properties, like cellular polarity and cell-cell contact by the epithelial cells. Vimentin, a cytoskeleton protein of 57 kDa, is a highly conserved and broadly expressed protein of the type III Intermediate Filament protein family. Vimentin expression is restricted to mesenchymal cells. Vimentin has gained much attention in cancer biology as a sanctioned marker of EMT, lately. EMT is a cellular reprogramming process in which the epithelial cells lose their cellular polarity, downregulation of the epithelial markers like E-cadherins and keratin, and acquires a mesenchymal phenotype that results in change of shape and shows increased motility of cells. The process of EMT is related to the development of resistance against certain chemotherapeutic drugs and vimentin expression has been observed to be upregulated in various tumor cell lines and tissues. Thus, we would like to study the role of vimentin in proliferation as well as sorafenib resistance in hepatocellular carcinoma.
In present study, the proteomic profiling of parental [HepG2 (P)] and sorafenib-resistant [HepG2 (R)] cells revealed the downregulation of vimentin in HepG2 (R) cells as compared to HepG2 (P) cells. This result was further confirmed at mRNA level by qRT PCR and at protein level by flow cytometry analysis. A study in 2016 by Yi Huo et al. also reported the similar pattern of downregulation of vimentin in cisplatin resistant ovarian cancer cells. Then, we investigated the effect of different doses of sorafenib on gene expression of vimentin and at 5 ┬ĄM sorafenib conc. there is significant downregulation of vimentin expression. Further, to evaluate significance of vimentin inhibition in sorafenib resistance, a well known inhibitor of vimentin was used that mediates its aggregation and disassembly. Withaferin A successfully lowered down the levels of vimentin in HepG2 (R) cells.
In HCC sorafenib resistance, ABCG2 is one of the prominent efflux pumps belonging to the family of ABC (ATP binding cassette) transporters, involved in efflux of sorafenib out of resistant cancer cells [25,26]. We analyzed the effect of withaferin A on ABCG2 expression in HepG2 (R) cells to understand its targeted action. In our study, withaferin A significantly decreased the levels of ABCG2 gene in HepG2 (R) cells which indicated towards the probable role of vimentin in the establishment of sorafenib resistance in HCC cells.
Studies reported about the cytotoxic effect of Withaferin A in cancer cells [27]. To evaluate its cytotoxic nature our both crystal violet as well as MTT assays has demonstrated that using increasing dose of withaferin A as well as in combination with sorafenib both resulted to decrease cell viability of both HepG2 (P) and HepG2 (R) cells significantly.
Thus, the modulation of vimentin may result in the shift in proliferation as well as sorafenib resistance in HCC. Hence, our results demonstrated that vimentin is an eminent marker as well as its significance as a potential therapeutic target to treat sorafenib resistance in HCC.
This study connotes that vimentin is crucial for cell survival as its inhibition resulted in cell cytotoxicity in both HepG2 (P) and HepG2 (R) cells. Nonetheless, vimentin expression in resistant HCC cells was observed to be lowered, the inhibition further has proved detrimental to the resistant cells. Further, deciphering the specific resistance mediators, the effect of withaferin A was checked on ABC transporter gene. For the first time, the present study suggesting that withaferin A leads to a decrease in the expression of ABCG2 gene in sorafenib-resistant HCC [HepG2 (R)] cells. The inhibitor, withaferin A, mediating its anti-cancer action through cell apoptosis significantly decreased the expression of vimentin as well as cell viability in a dose dependent manner. This observed shift in resistant cells towards a parental-cell profile further hints the effect of vimentin in sorafenib resistance as its inhibition with withaferin A as a combination therapy along with only current targeted drug, sorafenib.
However, our study has some limitations. All experiments were performed using only one human HCC cell line (HepG2) and it was not possible to perform in vivo experiments. Thus, future research in this area is required to completely understand the mechanism and validate this study.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Supplement1
Supplement1 Print
Print





