


INTRODUCTION
Cirrhosis is a progressive chronic liver disease characterized by diffuse fibrosis, severe interruption of intrahepatic venous flow, portal hypertension and hepatic insufficiency. Epidemiological studies indicate the existence of an increase in the prevalence of liver cirrhosis worldwide [1]. The natural evolution of cirrhosis is divided into two stages; a compensated cirrhosis, which is defined as the period between the onset of cirrhosis and the appearance of the first major complication of the disease and the decompensated cirrhosis, which defines the period following the development of ascites, gastrointestinal hemorrhage due to rupture of esophageal varices and hepatic encephalopathy.
Recent medical studies and the introduction of new and effective treatments of some etiological forms of cirrhosis, especially antivirals, have brought about important changes in our conception of the disease [2]. First, cirrhosis is no longer considered an irreversible progressive disease. In fact, decompensated cirrhosis can again be compensated cirrhosis or even return to pre-cirrhotic stages if the cause of cirrhosis is eliminated. Second, the initial list of organismic or systemic insufficiencies (liver, kidneys and brain) has been expanded and now includes the immune system, the intestine, the heart, the lungs, the adrenal glands, the muscles and the thyroid gland. Third, new mechanisms have been recognized that are implicated in the pathogenesis of cirrhotic complications, such as dysbacteriosis of the microbiota and systemic inflammation. Finally, it is increasingly clear that patients rarely die as a result of irreversible and terminal destruction of the liver. On the contrary, the cause of death in most patients is an acute deterioration of their clinical situation, favored by a triggering event, a syndrome called acute-on-chronic liver failure (ACLF)[3]. In critically ill patients, early diagnosis of ACLF could be important for therapeutic decisions.
FIBROGENIC PROCESS
Liver fibrosis is a healing process after hepatic injury with a dynamic and potentially reversible process, involving complex molecular and cellular mechanisms that lead to chronic activation of tissue remodeling and repair, however, if the damage persists, this process could advance to cirrhosis even to liver cancer [4].
Activated hepatic stellate cells are the main cell type responsible for liver fibrogenesis because they express, synthesize and secrete a broad spectrum of extracellular matrix proteins (collagen, glycoproteins and proteoglycans), during chronic liver injury [5]. Additionally, these cells produce pro-fibrogenic and proinflammatory cytokines, chemokines and growth factors, which are vital in the onset of fibrogenesis [5].
Moreover, activated hepatic stellate cells increase neutrophils survival by the production of granulocyte-macrophage-colonystimulating factor and interleukin (IL) 15 that exacerbate liver inflammation and increases reactive oxygen species, which are generated by neutrophils that promote hepatocytes damage. Apart from the possible effects on hepatocyte function, the increase in fibrosis significantly modifies the mechanics of blood flow in the liver leading to the formation of collateral vessels and arterial vasodilation progress, which eventually generates esophageal varices and ascites [6]. A high fructose and cholesterol diet activates early pro-fibrotic pathways before the development of proinflammatory signaling or insulin resistance [7]. In contrast, activation of natural killer cells inhibits hepatic fibrosis through the generation of interferon-╬│ that induces apoptosis and cell-cycle arrest of activated hepatic stellate cells [8].
Human bone marrow-derived mesenchymal stem cells inhibit the proliferation of activated hepatic stellate cells by the induction of apoptosis through two proteins, antiapoptotic Bcl-2, and proapoptotic Bax, which are key proteins to the mitochondria-mediated intrinsic apoptosis [9]. Furthermore, the generation of reactive oxygen species is intimately related to the hepatic fibrogenic process, where nicotinamide adenine dinucleotide phosphate (NADPH) functions as one of the main sources. It is considered that p47phox, an active functional component of the NADPH complex, plays a crucial role in its activity. The mesenchymal stem cells derived from the human bone marrow can restrict the activation of this oxidase by phosphorylation of p47phox, thus inhibiting its signaling pathway, which promotes peroxisome proliferatoractivated receptor ╬│ and inhibits the expressions of collagen ╬▒1 and ╬▒-smooth muscle actin [10].
Cirrhosis represents the final stage of the fibrogenic process, with a transition from an asymptomatic compensated stage, which is between cirrhosis appearance and the initial major complication to symptomatic decompensated stage [11]. Patients with compensated cirrhosis are characterized by having minor symptoms or not having any at all, however, if the etiological factor persists, hepatic damage and portal pressure could worsen. The second phase is decompensated cirrhosis, where variceal hemorrhage, jaundice, ascites and hepatic encephalopathy may occur, all of which are associated with poor short-term survival [3]. Once liver cirrhosis is present, there are several processes get underway like monocytes and lymphocytes interaction between the highly specialized sinusoidal endothelial cells,12 activation of the inflammasome driven by signaling by toll-like receptor (TLR) or interleukin-1 receptor (IL-1R), which leads to expression of inflammasome components such as NOD-like receptors, pro-caspase-1, pro-interleukin-1╬▓, and proIL18, as well as nitric oxide elevation [13].
DIFFERENCE OF ACUTE-ON-CHRONIC LIVER FAILURE ACORDING TO GEOGRAPHICAL AREA
In cirrhosis, there is a term called acute decompensation, which is characterized by severe hepatic disease development, being the primary cause of hospitalization with a high risk of death in the shortterm. Commonly these patients are usually diagnosed with ACLF [14].
There are different definitions of ACLF. The first one was described in 2002 by Jalan and Williams to explain how compensated cirrhotic patients show a severe deterioration in hepatic function during 2 to 4 weeks, associated with a precipitating event which leads to a severe clinical worsening with hepatic encephalopathy, jaundice, bacterial infection, ascites, gastrointestinal hemorrhage and/or hepatorenal syndrome [3]. There are disparities between the different committees that study the liver, according to the Asian Pacific Association for the Study of the Liver, ACLF is an acute hepatic damage that is manifested as coagulopathy and jaundice which gets complicated over a four-week period by ascites and/or encephalopathy in a patient with a chronic liver disease or cirrhosis and is associated with high mortality; though, bacterial infections are not considered hepatic insults. Patients with cirrhosis and known prior decompensation (jaundice, encephalopathy or ascites) who develop acute deterioration in clinical status ŌĆörelated or unrelated to precipitating eventsŌĆöhave acute decompensation, but not ACLF [15,16].
However, the North American Consortium for the Study of EndStage Liver Disease, states that patients with decompensated cirrhosis and bacterial infections that develop two organ failures: hepatic encephalopathy grades III-IV, septic shock, the need for mechanical ventilation or renal replacement therapy, present ACLF [3,17].
The European Association for the Study of the Liver (EASL), Chronic Liver Failure Consortium decided to apply a more pragmatic approach to define ACLF. According to the results of the EASL-chronic liver failure (CLIF) Acute-on-Chronic Liver Failure in Cirrhosis (CANONIC) study, ACLF is defined as a clinical syndrome characterized by acute decompensated cirrhosis that leads to multiple organ failure and a high mortality rate in the short term (mortality rate within 28 days Ōēź15%). In that study, the multiple organ failure was evaluated according to the sequential organ failure assessment score, which is widely used in critically ill patients but with certain modifications to accommodate the clinical setting of patients with cirrhosis which was called CLIF-sequential organ failure assessment score, with a later simplified version called CLIF-C organ failure score. This consensus also classified ACLF in three subtypes in relation to the chronic liver disease stage: Type A ACLF (chronic liver disease without cirrhosis), Type B ACLF (compensated cirrhosis) and Type C ACLF (non-compensated cirrhosis) (Table 1) [3,14,16-19].
This illness is widespread and associated with a very high mortality rate (28-day) compared to cirrhotic patients without ACLF.14 ACLF has a multifactorial process that includes acute events like immune deregulation, atypical plasma cytokine profile and continuous inflammation, among others. ACLF onset is related with events like hepatitis reactivation, alcohol or drug abuse and bacterial infection, which represent more than 50% hospitalization, being the main cause of decompensation in ACLF [20], therefore resulting in organ injury or multiple organ failure that can lead to death [20,21].
ACLF etiology is different according to the geographical area in which it is studied, for example in Western countries alcohol-related liver cirrhosis is the first cause of the disease, meanwhile in Asian countries 70-80% it is caused by the hepatitis B virus (HBV) infection.21 Globally, most young patients with ACLF have alcoholic cirrhosis [22].
TRIGGERING FACTORS AND PATHOGENIC MECHANISMS IN ACLF
Commonly, patients with acute decompensation or ACLF show an elevation in cell death markers, which increase with clinical severity compared with healthy patients or with stable cirrhosis. Especially from the pathophysiological point of view, since there is a high correlation with the activation of macrophages, the markers of inflammation and the oxidative stress; indicating that cell death is an important feature in ACLF [23]. Furthermore, there is a difference between the mechanism and the severity according to the etiology from which they are derived [23].
The process of decompensation itself can induce organ tolerance possibly because a previous hepatic lesion could produce cellular senescence [24] which makes the hepatocytes more resistant to apoptosis [25]. Patients who previously had not suffered from a decompensating event showed to have higher cell death markers and high mortality through mechanisms that have not been described yet [14,23].
ACLF triggers can be categorized into two groups: those that directly impact the liver and those that affect it secondarily to a non-hepatic lesion. In the first group, viral hepatitis, drugs and other hepatotoxic substances such as alcohol stand out. In the second group, variceal hemorrhage and bacterial infections show up as the main factors.
Cytoskeleton of hepatocytes and biliary tract cells is formed by cytokeratin 18 (CK18) filaments; therefore after apoptosis or necrosis CK18 is released to the bloodstream establishing as a cell death indicator that can be quantified by enzyme-linked immunosorbent assays, called M65. In apoptotic cells, the CK18 filaments are cleaved by caspases into a small polypeptide M30 that can be detected in serum by specific antibodies, which predict disease severity and survival of patients with ACLF. The rise of M30 and M65 were associated with other liver diseases severity such as cholestasis and chronic HBV infection. This small increase in polypeptides is related to the severity and progression of cirrhosis rather than the etiology of the disease [26]. Elevation of CK18 impairs liver function as shown in liver biopsies, due to a marked increase in apoptosis observed by terminal deoxynucleotidyl transferase dUTP nick end labeling staining. There is also an activation of neutrophil recruitment signals by IL-8 and anti-inflammatory agents that limit the immunological effect of cell death (IL-10, IL1RA, sCD163) [23,27]. The reactivation of HBV has been the major cause in which the ACLF is presented in most studies of the AsiaPacific region [28], presenting necrosis as the main characteristic [29].
Alcohol consumption as an acute precipitant in ACLF is associated with worse outcomes, such as advanced hepatic encephalopathy, elevation of serum creatinine and the presence of infection, in addition to high short-term mortality, when compared with other etiologies [30]. Also, patients with ACLF show a marked worsening in the circulatory disturbances present in liver cirrhosis. The mechanisms and pathophysiology have not been clearly determined, however, in a study with patients that present severe acute alcoholic hepatitis, blocking the tumor necrosis factor ╬▒ (TNF-╬▒) using infliximab produced a marked decrease in both portal pressure and the degree of hyperdynamic circulation [31]. On the other hand, the elevation of pathogen-associated molecular patterns has been related to liver damage by different pathophysiological mechanisms of decompensation, showing ballooning degeneration and cell death with a predominance of cholestatic infection [32,33].
When the type of organ failure correlates with mortality, cerebral and circulatory/pulmonary insufficiency is strongly associated in >95% of the cases. However, biliary nephropathy is an important toxic injury caused by elevated levels of bile acids and bilirubin, which is significantly common in patients with ACLF [22]. In addition, the number of organic failures is also correlated with mortality, increasing abruptly when there are more than two organic failures to 80% and increase up to 94% when there are more than 3 and 4 organic failures [28]. Among the available forecast scores, CLIF-C ACLF and Acute Physiology and Chronic Health Evaluation (APACHE) II present the best performance [30].
The physiopathological mechanisms that characterize the development of ACLF are not completely known, but they generally occur in the context of an intense systemic inflammatory response. In the CANONIC study, it was observed that patients with ACLF presented a marked elevation of C-reactive protein together with a rise of the leukocyte count. These findings led to the hypothesis that excessive systemic inflammatory response is a basis to explain the pathogenesis of ACLF [34].
ACLF INFLAMMATORY PROCESS
The majority of the altered cytokines in ACLF are related to chemotaxis and leukocyte migration, mainly macrophages and monocytes. This disturbance is not just associated with bacterial infections, given that cytokine levels were different in patients with bacterial infections out of ACLF when compared to patients with the illness [35].
A study conducted by Dr. Barroso shows that patients with ACLF had elevated levels of C-reactive protein and leukocytosis, because of an inflammatory event such as bacterial infections and severe alcoholic hepatitis in 57% of the patients, or to mechanisms that were not identified in the remaining 43%, which suggests an altered immune response that leads to an inadequate inflammatory response [22].
Patients with alcoholism have a neutrophil dysfunction, which increases the reactive oxygen species and alters phagocytosis together with a deficient bacterial death. Despite being in a proinflammatory state, they are more susceptible to infections, having a worse prognosis. In some cases, the infections are a consequence of ACLF instead than a cause [28].
On the other hand, patients with ACLF show higher levels in serum histone 4 compared to controls and it is not only significantly related to liver damage but also to the failure of other organs, disease severity and overtime mortality. Extracellular histones have diverse effects such as coagulation by platelet aggregation, white blood cell count and elevation of C-reactive protein in plasma, endothelial damage induction and production of cytokines (IL-1╬▓, IL-6, IL-8, IL-10, IL-18 and TNF-╬▒), which indicates an acute inflammatory response that in turn predisposes host to bacterial translocation and infection, worsening the inflammatory response [21,34]. Also, survival decrease in ACLF patients is related to the reduced human leukocyte antigen-D expression on monocytes, which increases the severity and risk of infection [15]. However, interferon-╬│, transforming growth factor ╬▓-1, IL-1╬▓ and IL-12 levels are normal [35].
Th9 cells are lymphocytes that produce IL-9 and IL-10, which migrate to the liver in response to acute damage and regulate the duration and the intensity of inflammation. In a study conducted by Dr. Zhijun SuŌĆÖs group, it was found that the percentage of Th9 cells was lower in patients with ACLF than in healthy patients [27]. However, patients who died showed a significant increase in IL-9 and IL-10, unlike patients with ACLF who survived and healthy controls, suggesting that other immune cells could secrete IL-9 and IL-10 [27].
Moreover, patients with ACLF have increased numbers of inmunoregulatory monocytes and macrophages that express MER receptor tyrosine kinase and suppress the innate immune response to microbes. The number of these cells correlates with disease severity and the inflammatory response. MER receptor tyrosine kinase expression positively correlates with IL-6, IL-10 and TNF-╬▒, which might suggest that these cytokines could be related to the induction of this in ACLF, therefore it could be said that it may serve as a prognostic marker for ACLF [20].
Neutrophil gelatinase-associated lipocalin is a LCN2 gene protective protein against bacterial infections given that it binds to the bacteria siderophores. The neutrophil gelatinase-associated lipocalin is another ACLF biomarker in plasma or urine, which can be used as a prognostic marker in patients with cirrhosis acute decompensation. As well it has been shown that hepatic LCN2 gene levels are increased in patients with ACLF and the expression of this gene is correlated with liver function parameters (bilirubin, albumin, international normalized ratio and the model for end-stage liver disease score) [36].
GENETICS OF ACLF
It has been pointed out that the pathogenesis of the ACLF may depend on the insult, the immune response, secondary infections or organ failure; however, the possibility of developing ACLF could be partially genetically predetermined [34,37].
In a genetic association study of candidate genes in twins, it has been suggested that host genetic factors are critical in determining the outcome of HBV infection [38,39]. The HBV infection depends on the interaction between the virus, the hepatocytes and the host immune response; even the same strains of HBV can lead to different levels of pathogenicity and severity, indicating an individual susceptibility to ACLF [37,40].
Although mortality in patients with ACLF-HBV is 30ŌĆō70%, most patients with chronic hepatitis B do not suffer from ACLF throughout their lives. This may be due to genetic variations in DNA sequences such as single-nucleotide polymorphism (SNP), which explains the individual biological differences and how they affect humans to develop a disease [41].
Moreover, a few studies showed that mutations of the basal core promoter, or in the core promoter of HBV genome may have different clinical implications [42,43]. In 2018 Zang et al reported that the detection of mutations in the HBV genome in the basal core promoter/core promoter region (A1846T and C1913A) is positively associated with the severity of liver disease and the risk of ACLF [44]. On the other hand, a genome-wide association study reported that 10 SNPs were associated with HBV-related to ACLF. The analysis showed that human leukocyte D antigen is related as the main locus for susceptibility to ACLF related to HBV. In addition, the responses of allelic CD4+ T cells related to human leukocyte D antigen may be important for the initiation and progression of ACLF related to HBV, which was replicated in four populations. Finally, it is proposed that one of the variants (rs3129859) could be used as a prognostic marker for the appearance, severity and survival of the ACLF related to HBV in patients with chronic HBV infection [41].
SNPs can also modify the immune response and the incidence of infections. The innate immune response serves as the first line of defense against pathogens and the malfunctioning of this lead to an increase in the incidence of bacterial and fungal infections [22]. The SNP rs2910164 of the miR-146a gene is involved in the regulation of innate immunity and is associated with the susceptibility to ACLF, therefore, a lower genetic risk of developing ACLF. Homozygous subjects for the SNP of the miR-146a gene had a higher survival rate and lower serum levels of TNF-╬▒, which decrease the induction of hepatocytes apoptosis and improve the severity of the disease [37].
Genetic polymorphisms of TLRs have been associated with an increased predisposition to infection in several populations; these receptors play a key role in the innate response and the recognition response to viral pathogens [45]. A case-control study (in a Chinese population) analyzed the correlation between TLR3 C1234T polymorphism and chronic hepatitis B related with ACLF. This receptor plays a key role in the inactive response and the recognition response to viral pathogens. This polymorphism was associated with increased risk of ACLF in chronic hepatitis B patients and may act as a potential marker for the prognosis [46]. TLR3 agonism may improve antibacterial responses and reduce infectious complications without having a detrimental effect on tissue repair processes [47]. Furthermore, being homozygous for the polymorphism TLR3 L412 was associated with a lower rejection rate for liver transplantation [48]. As well, increased expression of TLR4 in mononuclear cells from ACLF patients rises T cells activation, which leads to liver damage, indicating that TLR4 may play a role in the aggravation of ACLF disease [49]. Previously, the genetic polymorphisms of the TLR4 receptor have been particularly associated with D299G, however, at the polymorphisms of this receptor may not be associated in the predisposition to develop bacterial infections and therefore in the prognosis of cirrhotic patients with ascites (Table 2) [37,41,44,46,48,50].
CONCLUSION
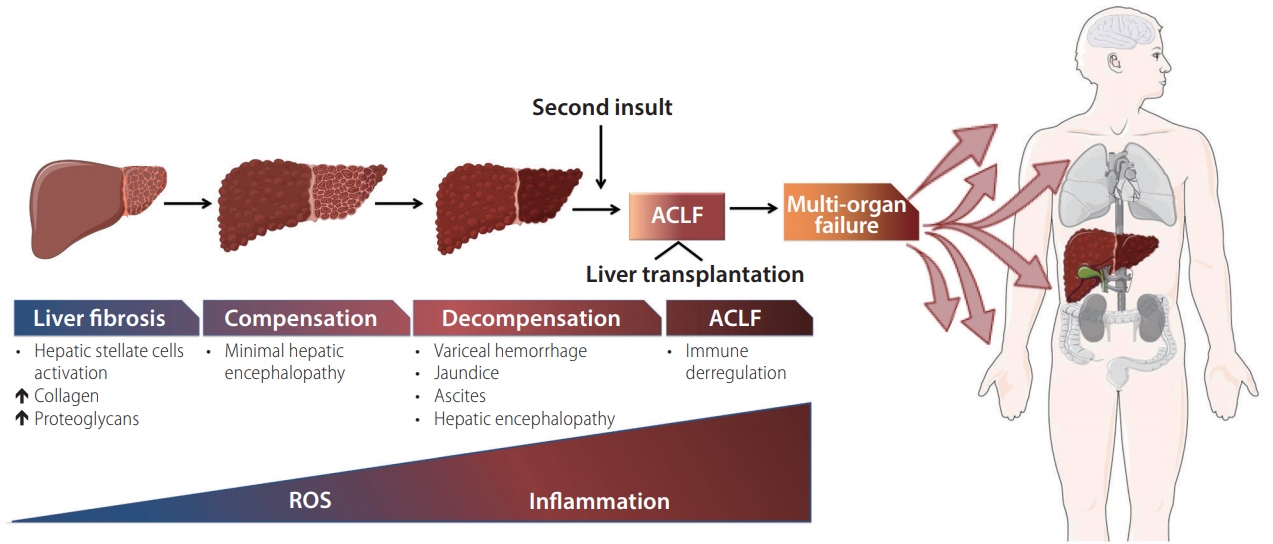
Chronic liver diseases and cirrhosis are intricate pathological stages with a heterogeneous pathophysiology in which genetic factors of the host and external triggers interact and culminate in a liver failure that leads to multiple clinical complications with a high rate of morbidity and mortality (Fig. 1).
During the last decade several studies have been carried out on the progression of liver damage through the interaction between hepatic parenchymal cell dysfunction and the immune system in the pathological process. These harmful processes can generally lead to the development of fibrosis, however, there are some cases where it does not occur in such a way. This point encourages us to continue basic and clinical researchs to elucidate the multiple pathways involved in liver damage and ACLF, leading to the discovery of new treatments that will make acute decompensation preventable and even reduce mortality.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print





