

INTRODUCTION
Hereditary hemorrhagic telangiectasia (HHT), also known as Osler-Weber-Rendu disease, is an autosomal dominant multiorgan disorder caused by a genetic defect encoding a protein that binds transforming growth factor, which results in abnormal vascular remodeling in the form of fibrovascular dysplasia with multiple telangiectasias.1 HHT is caused by mutations in one of two genes that have led to its subclassification into type 1 or 2 disease in most cases. HHT type 1 disease, the disease-causing gene is located on chromosome 9, encoding endoglin (ENG).2 In HHT type 2, the disease-causing gene is located on chromosome 12, encoding activin receptor-like kinase 1 (ACVRL1).3 HHT is characterized by the classic triad of mucocutaneous telangiectasias, recurrent epistaxis, and familial occurrence.4 Multiple telangiectases consisting of thin-walled, dilated vascular channels with arteriovenous malformations may involve mucocutaneous tissue, gastrointestinal tract including the liver, lung, and brain.5 The condition usually manifests in adults with epistaxis, hemoptysis, or both, and hepatic involvement is typically diagnosed 10-20 years after the first telangiectasias appear.6,7 The diagnosis of HHT is established when any three of the following criteria are present: (1) spontaneous and recurrent epistaxis; (2) multiple telangiectasias at characteristic sites, including lips, oral cavity, fingers, and nose; (3) presence of internal lesions such as gastrointestinal telangiectasia, pulmonary, hepatic, cerebral, and spinal arteriovenous malformations (AVMs); and (4) family history in the first-digree relative.8
Although the genotypic-phenotypic correlations are not yet fully defined, disease severity is more pronounced in HHT type 1 than in HHT type 2 with earlier age of onset for epistaxis and hepatic involvement were observed predominantly in HHT type 2.9 We present here a case of HHT type 2 with hepatic involvement with hepatocellular carcinoma (HCC) and confirmed by the mutation in the ACVRL1. Transarterial chemoembolization (TACE) for the unresectable HCC was not effective in this case and even risky procedure when HCC is associated with HHT.
CASE REPORT
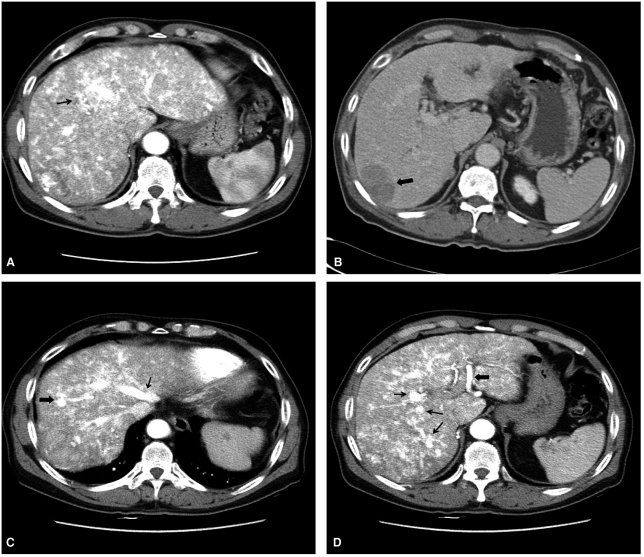
A 68-year-old man was referred for evaluation of a hepatic mass. He had history of hepatitis B virus carrier for 20 years and liver cirrhosis for 3 years. He has been taking an angiotensin receptor blocker and digoxin due to hypertension and heart failure. He has suffered from repeated episodes of epistaxis from his teens and gastrointestinal bleeding from his mid-thirties. All of his five children also had the history of recurrent epistaxis. Also his sister was suffering from the sequelae of cerebrovascular accident. On physical examination, several telangiectatic macular lesions were found on his arms and trunk (Fig. 1A, 1B). The laboratory work-up at first visit revealed the following: WBC, 4.6├Ś103/mm3; platelet, 172├Ś103/mm3; hemoglobin, 12.6 g/dL; aspartate aminotransferase (AST), 42 IU/L; alanine aminotranferase (ALT), 20 IU/L; hepatitis B virus surface antigen, positive; hepatitis B surface antibody, negative; anti-hepatitis C virus antibody, negative; and alpha-fetoprotein (AFP), 345 ng/mL (normal, 0-20). Other laboratory results including bleeding time, coagulation time, prothrombin time and activated partial thromboplastin time were normal. Gastroduodenal endoscopy disclosed a telangiectasia at the lesser curvature in mid-body of the stomach (Fig. 1C). Liver dynamic CT (LDCT) revealed multiple HCCs in the right lobe of liver and there was no evidence of distant metastasis or vascular invasion. The HCC lesions showed heterogenous wash-out compared to well enhancing round homogenous AVMs in delayed phase of LDCT (Fig. 2).
Liver transplantation was recommended, however, the patient refused it due to economic problem. TACE was performed in three consecutive sessions, but lipiodol uptake in the tumor mass was very poor at every session. HCC progressed after each TACE session. Fever developed up to 38Ōäā, and AST and/or ALT increased more than two times of baseline following the first two TACE sessions.
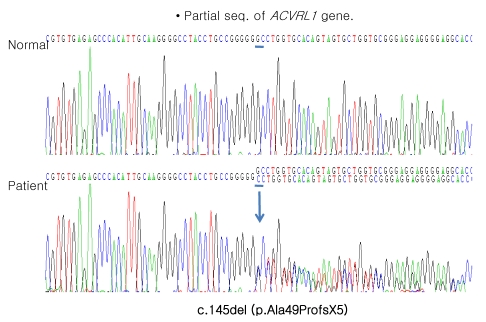
Because of unusual TACE effect, we reviewed all the radiologic images of the patient and found an enlarged tortuous hepatic artery, multiple arteriovenous malformations, and early visualization of hepatic veins in LDCT (Fig. 2) and hepatic angiography (Fig. 3). Based on these findings and positive family history, he was diagnosed as Osler-Weber-Rendu disease and genetic tests for the ENG and ACVRL1 were performed. Patient's genomic DNA was isolated from peripheral blood using the QuickGene DNA blood kit (Fujifilm, Tokyo, Japan) and BigDye Terminatore V3.1 Cycle Sequencing Ready reaction kit (Applied Biosystems, Foster city, CA, USA). Fourteen and ten exons and their intronic flanking sequences of the ENG and ACVRL1, respectively, were amplified by polymerase chain reaction with appropriate primers (Table 1 and Fig. 4). The molecular genetic analysis for the whole exons with their flanking sequences of the ENG was negative. However, the patient harbors a deletion mutation, c.145del (p.Ala49ProfsX5), in the ACVRL, the diagnosis of HHT type 2 with hepatic involvement was confirmed by the results of genetic study. He is waiting for deceased donor liver transplantation for the treatment of HCC.
DISCUSSION
HHT or Osler-Weber-Rendu disease is a rare systemic fibrovascular dysplasia with autosomal dominant transmission, characterized by recurrent epistaxsis, mucocutaneous telangiectasis, and visceral AVMs.1 It has an estimated prevalence of 1 in 5,000-10,000. Recurrent epistaxis is often the first and most common manifestation.4 The dilated post capillary venule fuses with an arteriole bypassing the capillary system and resulting in an arterio-venous communication.5
HHT is caused by the mutation of the genes involved in the transforming growth factor-╬▓ signaling pathway which plays an important role in the formation of vascular endothelium.8 Two major types of HHT (HHT type 1 and type 2) are due to mutations of ENG and ACVRL, respectively. The ENG is located on the long arm of chromosome 9 (9q33-34) and determine the HHT type 1 disease, which correlates with a more critical pulmonary involvement and earlier age of onset for epistaxis.10 The ACVRL1 located on the long arm of chromosome 12 (12q13) indicate the HHT type 2 disease, with a predominance of liver involvement.2 Although the genotypic-phenotypic correlations are not yet fully defined, it appears that the liver involvement is more prevalent in HHT type 2. Our case also revealed to have the deletion mutation in the ACVRL, and the diagnosis of HHT type 2 was made. He showed epistaxsis, mucocutaneous telangiectasis and predominant hepatic AVMs with family history.
Hepatic involvement occurs in 30% to 73% of patients with HHT.11 However, most of them are asymptomatic. Symptomatic liver involvement is quite rare and symptoms occur in about 8% of the patients with HHT and liver vascular malformations.3 Patients with hepatic involvement have hyperdynamic circulation resulting from arteriovenous shunting, portovenous shunting, or both, and may develop high-output cardiac failure. Other complications include extrahepatic shunts, portal hypertension, hepatic portosystemic encephalopathy, biliary ischemia, cholangitis, and liver failure.6,7 The development of portal hypertension is mostly the result of shunting of blood from the hepatic artery to the portal vein. Perfusion abnormalities and tissue ischemia in the liver parenchyma can induce fibrosis around the abnormal vessels and hepatocellular regenerative activity. The combination of fibrotic telangiectases and nodular regenerative hyperplasia makes it hard to diagnose true hepatic mass lesions.11
The LDCT can demonstrate an enlarged hepatic artery, arteriovenous shunt, and early draining hepatic vein in the liver.12,13 Hepatic arteriography can be the best method of screening to characterize the liver vascular malformations.
The diagnosis of HCC is not easy in the background of the liver involved by HHT, because typical arterial enhancement and delayed washout pattern of HCC in dynamic CT scan could be masked by widespread AVMs and focal nodular hyperplasia common in HHT.10 In our case, the diagnosis of HCC was possible due to typical radiologic features and elevated AFP level without liver biopsy.
HCC associated with HHT were rarely reported. Two cases reports have been published since 1975.14,15 It has been suggested that development of HCC in HHT patients might be due to increased liver cell turnover resulted from 'over-nourishment' of liver sinusoids by hepatic AVMs. So far, the causal relationship between HHT and HCC development is not clear.
TACE is one of effective treatment modalities especially in patients with unresectable HCC.16 However, the presence of AVMs due to HHT could be an important barrier of effective TACE because blockade at feeding vessel in tumor is frequently impossible due to presence of AVMs. Usually the intrahepatic biliary tree obtains nearly all of its blood from the hepatic artery, so the presence of natural shunt from the hepatic artery to the hepatic or portal vein will result in biliary ischemia.17 In this situation, the hepatic artery embolization might be a quite risky procedure.3 In this case, three sessions of TACE were performed for the treatment of HCC, but those were ineffective and more severe deterioration of liver function was observed after TACE.
No treatment is recommended for HHT patients with asymptomatic liver involvement. In HHT patients with symptomatic liver disease, treatment can be divided into symptomatic treatment for the specific complications and management aimed at reducing the shunt by surgical ligation or transarterial embolization.3 However, the hepatic artery embolization/ligation should be considered mainly in non-transplant candidate who failed maximal medical therapy due to the risk of necrotizing cholangitis and liver failure. Orthotopic liver transplantation has been proposed as the only definitive curative option for the symptomatic liver involvement in HHT.18
In conclusion, the diagnosis of HCC in the liver involved by HHT can be difficult due to its vascular malformation and regenerative nodular hyperplasia. TACE is ineffective in patients with HCC associated with HHT, therefore liver transplantation should be considered as the first-line treatment modality. We report here a case of HHT, presenting with HCC which developed in the background of multiple intrahepatic AVMs.



 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print




