


Dear Editor,
Neonatal hemochromatosis (NH) is a severe, progressive, lifethreatening disorder that leads to hepatic failure during the first weeks of life and is associated with massive iron deposits [1-7]. Although its exact etiology remains to be better clarified, it has been hypothesized that maternal alloimmunity [1] and mitochondrial dysfunction [2] might play important roles in this disease.
This condition differs from the main disorders that present with liver iron deposits, such as HFE-related hemochromatosis (autosomal recessive disorder caused by biallelic mutations in HFE gene in which iron deposits lead to adult-onset liver, heart, testicular and pancreatic involvement; OMIM#235200), juvenile hereditary hemochromatosis (autosomal recessive condition associated with biallelic mutations in HJV and HAMP genes and clinically similar to HFE-related hemochromatosis, but earlier onset and fast progression; OMIM#602390 and 613313), TFR2-related hereditary hemochromatosis (autosomal recessive condition associated with biallelic mutations in TFR2 gene and clinically similar to HFE-related hemochromatosis, but earlier onset and slow progression; OMIM#604250) and ferroportin (SLC40A1)-related iron overload (autosomal dominant condition associated with monoallelic mutations in SLC40A1 gene; OMIM#606069).
Our male infant was born at term (39 weeks of gestation, birth weight: 2,615 g), after an uneventful pregnancy. His parents were healthy, caucasian, non-consanguineous and have not reported any familial history of liver, genetic, metabolic or syndromic conditions. His general conditions deteriorated after delivery and he required intensive care unit admission for anemia, bleeding, hypoglycemia, hypoactivity and unconjugated hyperbilirubinemia. During the following days, he developed recurrent hypoglycemia, cholestatic jaundice, hepatomegaly and hepatic dysfunction. He was then transferred to our tertiary-care unit at the 33rd day of life.
Laboratory tests demonstrated anemia (hemoglobin 9.1 g/dL; hematocrit 26.3%), thrombocytopenia (platelet count: 49,000/╬╝L), abnormal synthetic liver function (International Normalised Ratio [INR]>10), slightly elevated liver enzymes (alanine aminotransferase [ALT] 65 U/L; aspartate aminotransferase [AST] 41 U/L), elevated serum ferritin (1,487 ng/mL) and abnormal iron profile (serum iron 142 ╬╝g/dL; transferrin 103 mg/dL). Abdominal ultrasonography showed signs of liver disease and minimal-volume ascites. Echocardiography showed atrial septal defect and mild pulmonary valve stenosis.
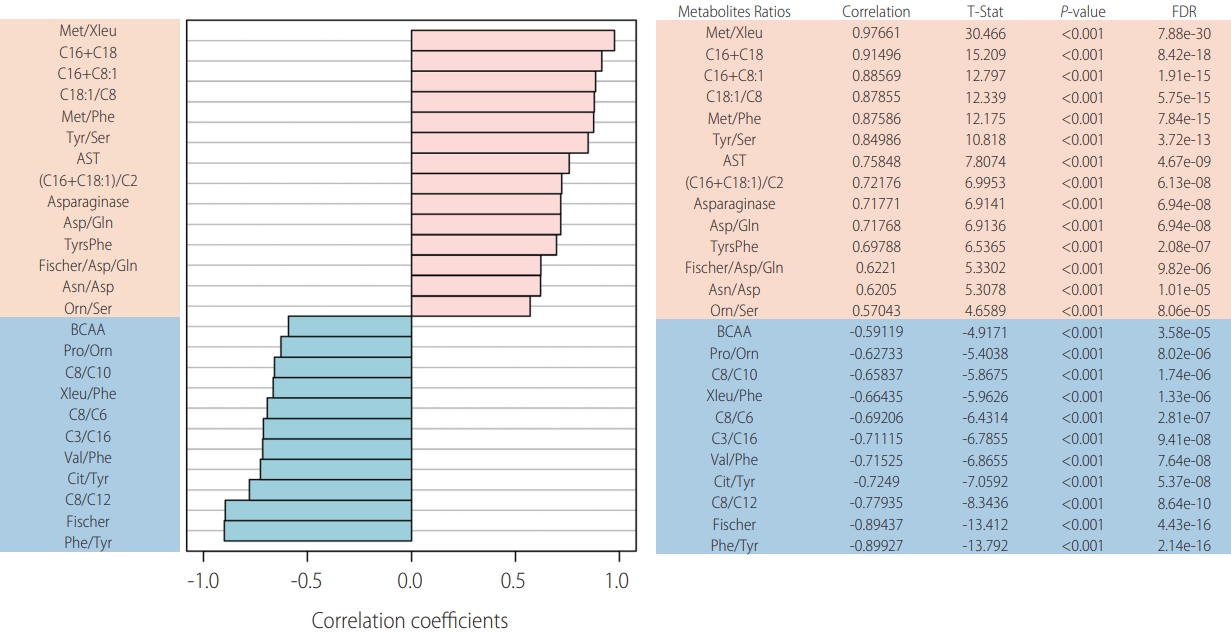
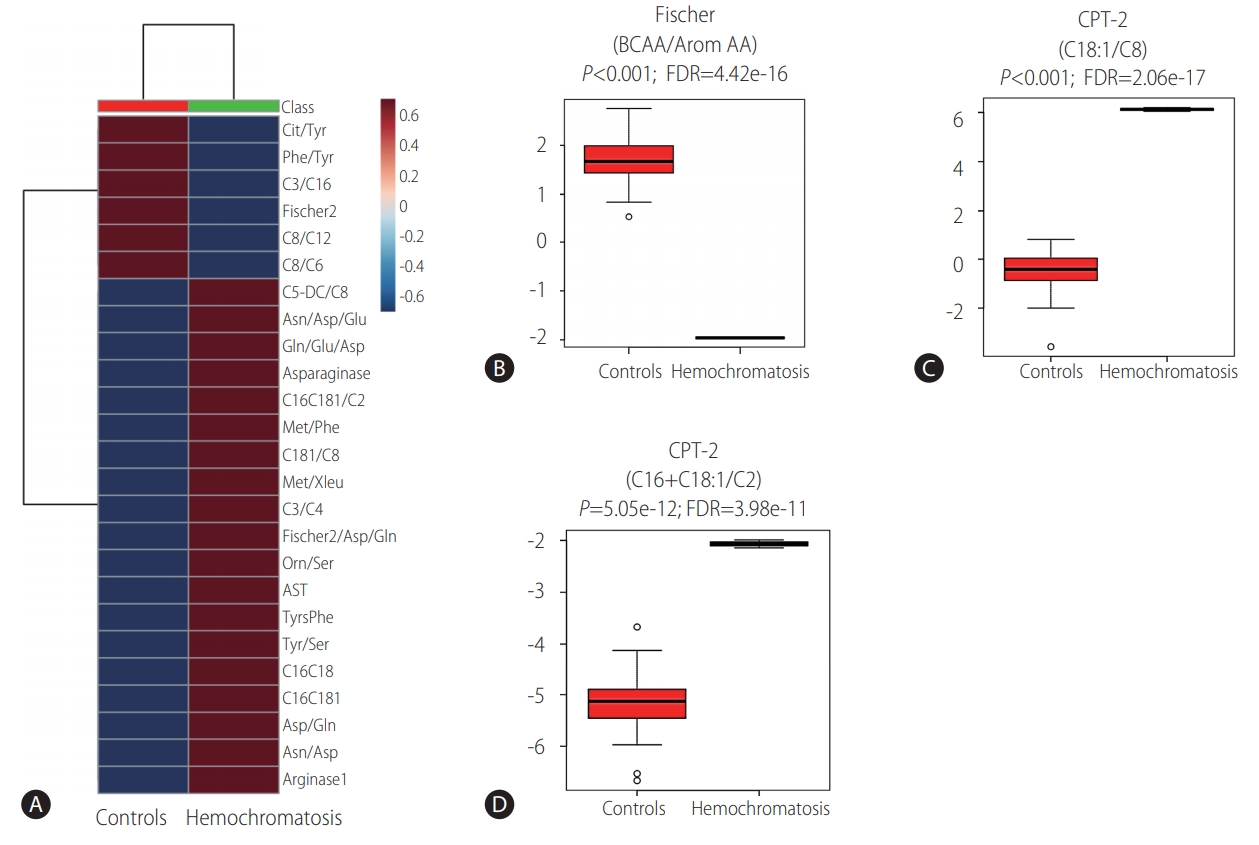
Laboratory workup included comprehensive metabolic investigation. Mass spectrometry analysis of acylcarnitine and amino acid profiles demonstrated unusual patterns when compared to 44 internal controls (Fig. 1). Statistically significant high ratios of methionine and tyrosine (methionine [Met]/leucine+isoleucine [Xleu], Met/phenylalanine [Phe], tyrosine [Tyr]/serine [Ser] and Tyr/Phe), which are non-specific markers of acute hepatic lesion, along with low levels of branched-chain amino acids (BCAA, XLeu [leucine+isoleucine]/Phe, valine [Val]/Phe) and Fisher ratio (BCAA/aromatic amino acids) were observed. Succinylacetone, which is considerably elevated in tyrosinemia type I, was normal and ruled out this condition. Impairment of urea cycle, a pathway highly dependent on liver metabolic capability, was also noted, with low levels of citrulline (Cit/Tyr) and high levels of ornitine (Orn/Ser) ratios. Moreover, acylcarnitine analysis demonstrated statisticallysignificant elevation of long chain species (C16+C18, C18:1/octanoylcarnitine [C8]) resembling carnitine palmitoyltransferase II (CPT-2) deficiency (OMIM: 600650), an inherited disorder of mitochondrial long-chain fatty acid oxidation (Fig. 2).
Investigation also included next-generation sequencing (NGS) panel (Illumina HiSeq based sequencing platform followed by Nextera Rapid Capture Mendelics Custom Panel V2 capture kit and in-house bioinformatics workflow), focused on treatable metabolic disorders, which resulted normal. Target genes contained in NGS panel are available in Supplementary Material. Unfortunately, DGUOK and POLG genes, respectively correlated with Mitochondrial DNA depletion syndrome 3 (hepatocerebral type) (OMIM: 251880) and Mitochondrial DNA depletion syndrome 4A (Alpers type) (OMIM: 203700), as well as genes related to latteronset hemochromatosis (HFE, HJV, HAMP, TRF2 and SLC40A1) were not studied due to the absence of available samples.
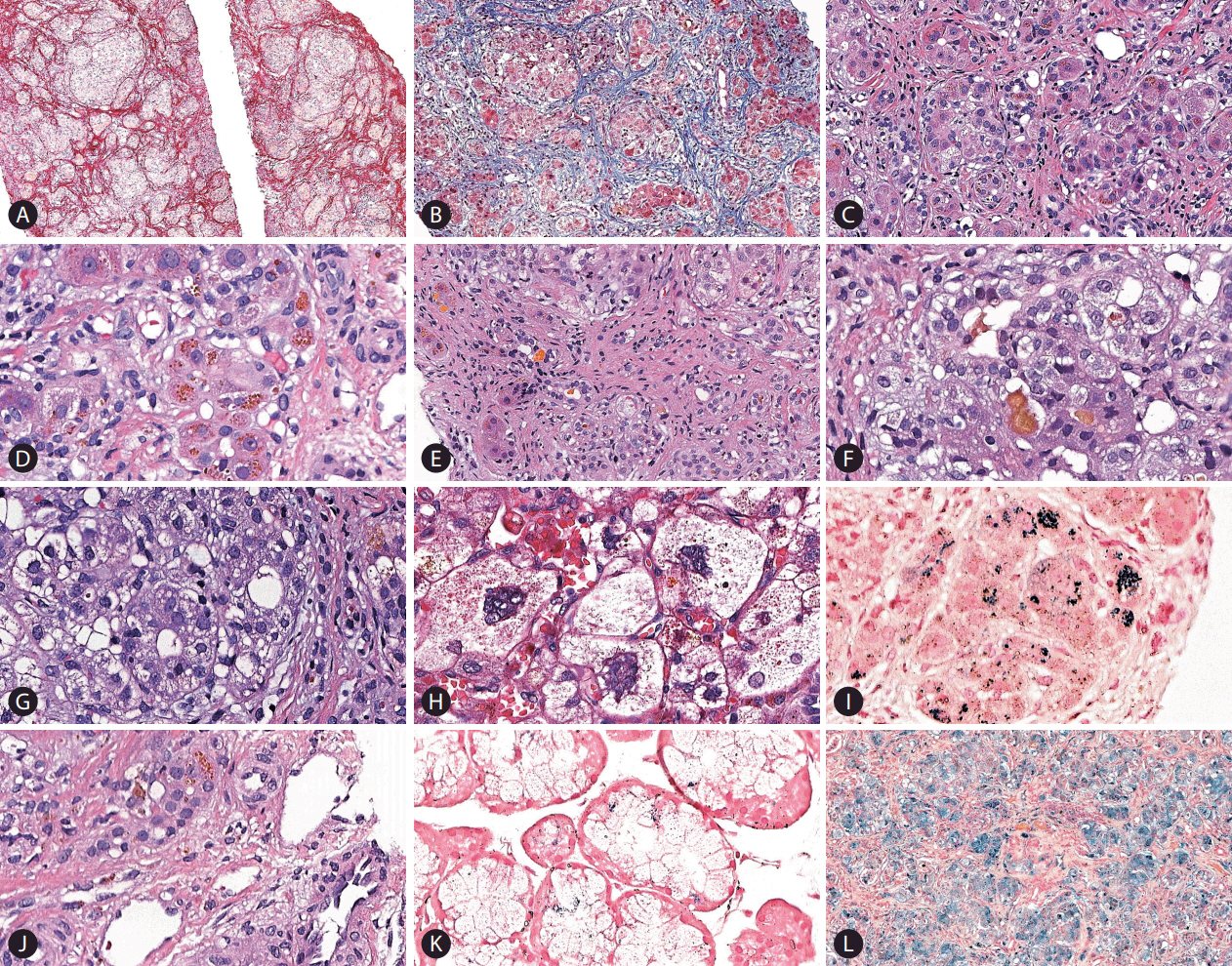
Liver biopsy (Fig. 3) revealed neonatal hepatitis syndrome characterized by micronodular cirrhosis (Fig. 3A), diffuse lobular fibrosis (Fig. 3B, C) associated with previous loss of hepatocytes, cholestasis (Fig. 3F), ductular reaction (Fig. 3E) with occasional inspissated bile (non-obstructive pattern), pseudoacinar transformation of hepatic plates (Fig. 3G), multinucleated giant hepatocytes, hepatocellular ballooning degeneration (Fig. 3G), extramedullary haematopoiesis and coarsely granular siderosis (Fig. 3D, I) especially in hepatocytes (grade 3) and even in rare bile ducts (Fig. 3J). A biopsy of oral mucosa showed abnormal iron deposition in minor salivary glands (Fig. 3K). Hepcidin expression was not studied because immunohistochemistry antibodies for this molecule are not available in our department.
The patient presented clinical complications during the investigation that delayed liver biopsy and the diagnosis of hemochromatosis. During the diagnostic workup of liver and metabolic profiles, the patient developed fast, progressive hepatic dysfunction with severe coagulopathy, anasarca and renal failure. This unexpected clinical picture strongly suggested acute liver failure, as his Pediatric End-Stage Liver Disease (PELD) score was 54 at that moment. Multidisciplinary evaluation of the level of hepatic dysfunction and the fast progression of the disease considered the case severe enough that liver transplantation could not be abrogated any further. Unfortunately, combination of exchange transfusion and intravenous immunoglobulin had not been available at that time at our service. The infant underwent urgent orthologous liver transplantation at 4 months and 22 days of age and received a segment graft (240 g, approximately 4.3% of his weight) from his mother (liver segments II and III). During the procedure, he developed uncontrolled hemorrhage and the surgery needed to be postpone 24 hours to be concluded. Histopathological examination of the liver explant demonstrated many microscopic findings similar to the core biopsy described above, including a shrunken liver due to cirrhosis and extensive parenchymal fibrosis, bilirubinostasis, giant cell transformation (Fig. 3H), remarkable siderosis (Fig. 3L), as well as scarce regenerative nodules. After 54 days from the procedure, the infant developed sepsis as complication of post-transplant immunosuppression and passed away at the age of 6 months.
Neonatal hemochromatosis is a rare, high-mortality condition associated with severe liver involvement of overt onset within the first weeks of life and extrahepatic tissue siderosis [3]. It is one of the main causes of neonatal liver failure. Serum ferritin levels are elevated to above 800 ng/mL in over 95% of patients and the demonstration of extrahepatic iron deposits is key to the diagnosis [3]. Therefore, the demonstration of hepatic and extra-hepatic iron deposits in this case established the definite diagnosis. Literature presents only a few sporadic reports, then its real incidence is not known. Liver transplant has been already performed before for treating this condition [8,9].
It has been postulated that maternal alloimmune response might play a role in neonatal hemochromatosis by altering fetal liver hepcidin expression, leading to iron overload and deposits in several tissues [4]. Hepcidin is regulated in and secreted by hepatocytes in the liver [10]. This mechanism implies that iron deposits are not the cause, but the consequence of liver dysfunction. This mechanism differs from the pathophysiology of the other iron-deposit conditions stated above. The main support for this theory relies on the greater survival rates of patients treated with the combination of exchange transfusion and intravenous immunoglobulin, focusing on removing offending antibodies [6]. Unfortunately, intravenous immunoglobulin was not an option in our service at that time. As our patient developed severe liver failure and consequently postponing liver transplantation to try other therapies was not an option.
Although the literature report that outcomes for liver transplant in neonatal hemochromatosis were not significantly different when compared to patients with other causes of hepatic failure, neonatal liver transplantation is always challenging once these patients are often critically ill with impaired multisystemic function and usually have low weight [7,8].
Enterocyte iron stored in ferritin is internalized into the bloodstream through the basolateral iron exporter ferroportin, which facilitates the transport and loading of iron into transferrin, the iron transport protein in plasma [9]. Serum hepcidin binds ferroportin and play an important role of negative regulator of total body iron influx [9]. Pathogenic mutations in HFE, HJV, HAMP and TRF2 genes are associated to the latter forms of hemochromatosis by the result of inducing low levels of hepcidin expression, while pathogenic mutations in ferroportin gene (SLC40A1) makes it insensitive to hepcidin regulation [9]. It is important to note that in these forms of hemochromatosis, liver dysfunction is acquired over time secondarily to iron overload. Although these genes were not studied in NGS panel making it not possible to rule out the presence of pathogenic mutations, the typical clinical progression of these conditions is usually different. Therefore, we believe that HFE, HJV, HAMP, TRF2 or SLC40A1-related forms of hemochromatosis are improbable to explain the etiology of our patient, although we cannot completely rule out this possibility.
Mitochondrial dysfunction, particularly mitochondrial DNA depletion syndrome, which is associated with biallelic DGUOK gene mutations and leads to impairment of energy metabolism, has also already been implicated as a possible cause of neonatal hemochromatosis [7]. It is not clear to us if the metabolic pattern found in our patient was exclusively secondary to hepatic failure or part of pathophysiology that led to his clinical phenotype. Literature lack data on this issue and the exact relation between these findings remains to be determined.
The major limitation of the present study is that the monogenic forms of hemochromatosis, the main nuclear genes that control mitochondrial function and mitochondrial DNA were not studied in the NGS panel. Therefore, we cannot conclude about the exact etiology of hemochromatosis in this patient.
The carnitine palmitoyltransferase II (CPT-2), the product of CPT2 gene, is located on the inner mitochondrial membrane and is an essential enzyme for fatty acid oxidation, a complex, multistep mitochondrial process that metabolizes fat into energy. Longchain fatty acids must be attached to carnitine to enter mitochondria and CPT-2 plays an important role by removing carnitine from this group of acylcarnitines and attaching coenzyme A (CoA), which is a necessary step for them to undergo beta-oxidation [10].
CPT-2 deficiency (OMIM#600649, 608836, 255110) is an autosomal recessive condition caused by biallelic pathogenic mutations in CPT2 gene and clinically associated with varying degrees of seizures, liver failure, cardiomyopathy, arrhythmia, non-ketotic hypoglycemia, myopathy, episodes of rhabdomyolisis, among other symptoms. The CPT2 gene was included in the NGS panel of the patient and its study did not demonstrate pathogenic mutations, ruling out primary CPT-2 deficiency. In primary CPT-2 deficiency, acylcarnitine profile demonstrates decreased levels of free carnitine (C0) and increased levels of long chain sterificated carnitines, specially palmitoyl-carnitine (C16), stearoyl-carnitine (C18) and vaccenyl-carnitine (C18:1).
Acylcarnitine profile of our patient demonstrates signs of impairment of mitochondrial long-chain fatty acids oxidation. As our study could not elucidate the exact etiology of the systemic, massive iron deposits, we cannot conclude whether mitochondrial dysfunction was primary (as a result of mutations in genes that control mitochondrial function, such as POLG, DGUOK and even mutations in mitochondrial DNA), secondary to alloimmunization, a pattern found in some cases of hepatic failure or, less likely, associated to severe forms of HFE, HJV, HAMP, TRF2 or SLC40A1-associated hemochromatosis. Once this is the first report of mitochondrial long-chain fatty acids oxidation impairment in a patient with neonatal hemochromatosis, other studies will be necessary to clarify whether this metabolic pattern is part of pathophysiology of this condition or has a more complex relation with it.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Supplement
Supplement Print
Print





