


A decade of liver organoids: Advances in disease modeling
-
Yue Liu1,2,*, Jian-Ying Sheng1,*, Chun-Fang Yang1,*, Junjun Ding2,3, Yun-Shen Chan1

- Received November 29, 2022 Revised January 13, 2023 Accepted February 28, 2023
- ABSTRACT
-
Liver organoids are three-dimensional cellular tissue models in which cells interact to form unique structures in culture. During the past 10 years, liver organoids with various cellular compositions, structural features, and functional properties have been described. Methods to create these advanced human cell models range from simple tissue culture techniques to complex bioengineering approaches. Liver organoid culture platforms have been used in various research fields, from modeling liver diseases to regenerative therapy. This review discusses how liver organoids are used to model disease, including hereditary liver diseases, primary liver cancer, viral hepatitis, and nonalcoholic fatty liver disease. Specifically, we focus on studies that used either of two widely adopted approaches: differentiation from pluripotent stem cells or epithelial organoids cultured from patient tissues. These approaches have enabled the generation of advanced human liver models and, more importantly, the establishment of patient-tailored models for evaluating disease phenotypes and therapeutic responses at the individual level.
- INTRODUCTION
- INTRODUCTION
The liver is a complex organ that is vital for regulating metabolism, energy storage, and detoxification in the human body [1]. The liver is the largest internal organ, and it mainly comprises hepatocytes that are central to the performance of most of its functions [2,3]. Other cell types in liver tissue include parenchymal cholangiocytes and nonparenchymal liver cells (NPCs): liver sinusoidal endothelial cells, hepatic stellate cells (HSCs), Kupffer cells (KCs), and other immune cell types. Each cell type plays different roles, and together, they maintain the structural integrity of liver tissue during homeostasis and make repairs when needed due to injury or disease [4,5]. These cells are spatially organized into lobules and form channels, including the sinusoid and bile canaliculi networks, that are essential for transporting nutrients into the liver and exporting metabolites and bile into the digestive system. In addition to the loss of hepatocytes, disruption of these liver structures and cellular interactions often occurs during drug-induced liver injury (DILI) and liver disease, resulting in the loss of liver function.Chronic liver disease (CLD) remains a leading cause of death and morbidity worldwide [6]. CLD is the major contributor to liver injury, fibrosis development, and the eventual progression to cirrhosis and primary liver cancer (PLC). Historically, the main etiology for CLD is viral hepatitis. However, with the development of antiviral treatments and vaccinations, a pandemic of obesity, and increased alcohol consumption, the primary emerging CLD etiologies are nonalcoholic fatty liver disease (NAFLD), alcoholic liver disease (ALD), and DILI [6,7]. Due to the complex interplay among the cellular components of the liver in varying disease conditions, it has been challenging to construct a robust human model for understanding disease manifestations and treatment responses [8,9]. Most human liver disease models have focused on the ways in which diseases and chemical agents induce hepatocyte injury and cell death. However, apart from those cellular changes, remodeling of the hepatic extracellular matrix (ECM) also plays an integral role in disease progression. Fibrogenesis is a common pathophysiological feature in injured and diseased livers, and it involves extensive ECM remodeling by activated HSCs [10]. These cells secrete fibril-associated collagens and matrix metalloproteinases that gradually remodel the soft and perforative endothelial/epithelial basal lamina, making it rigid and obscure. To accurately reflect disease progression, it is essential for research models to capture such ECM changes as they are driven by interactions among various cell-types.Scientists have long used three-dimensional (3D) culture methods to create human organ models that mimic in vivo niches and promote multidimensional cell–cell and cell–environment interactions [11,12]. In recent years, two cell culture platforms for generating such 3D cellular models have been increasingly adopted by laboratories across various research fields: pluripotent stem cell (PSC)-derived organoids and tissue-derived organoids. Since their discovery, PSCs have been an invaluable cell source for creating almost all cell types in the human body [13,14]. PSCs have a limitless self-renewing capacity and the ability to mimic embryogenesis and give rise to adult tissue cell types, and they are highly amenable to gene editing, so they are a preferred starting cell source for generating human organ models. One of the first 3D approaches to initiate differentiation into various germ layers is embryoid body formation, in which cells of all three germ layers begin to form [15]. It remains an initiation step in many differentiation protocols for PSC-derived organoids. To date, organoids resembling the brain [16], lung [17], kidney [18], stomach [19], intestines [20], pancreas [21], and liver [22,23] have been generated. The advent of human induced pluripotent stem cell (iPSC) reprogramming technology, which enables the generation of patient-specific PSC cells, has further accelerated the adoption and utility of the PSC-derived organoid platform [24].The ability of isolated adult mammalian cells to reassemble themselves into functional tissues when embedded in a culture matrix was first demonstrated using a 3D culture of epithelial cells derived from mammary glands and lung alveoli [25,26]. Both studies highlighted the ability of basement membrane matrix secreted from Engelbreth-Holm-Swarm sarcoma (commonly known as Matrigel) to support 3D human cell culture and the marked improvements in functioning shown by the 3D cultured cells compared with their twodimensional (2D) counterparts. In a landmark study by the Hans Clevers group, a single adult crypt stem cell was shown to generate functional intestine crypt organoids in Matrigel culture [27]. Specifically, those authors showed that by supplementing the cultures with growth factors and small molecules that modulate essential signaling pathways supported by the tissue niche, adult stem cells marked by Leucine-rich repeat-containing G-protein coupled receptor 5 (LGR5) could be cultured and propagated for an extended period. Clevers’s group and others quickly showed that by manipulating culture media to activate essential signaling pathways that support progenitor cells in other tissue types, the platform could be adopted to derive organoid cultures from other organs, including the liver [28-30]. Remarkably, the tissue-derived organoid platform is also highly suitable for deriving cancer cells from tumors of various indications. These 3D cancer cell cultures, commonly called tumoroids, have facilitated a breakthrough in deriving patient-specific cancer cells for colon, pancreatic, liver, ovarian, and nasopharyngeal cancer [31-35].Human models, including immortalized hepatic cell lines, PSC-derived hepatocyte-like cells, and primary human hepatocytes (PHHs), have been widely used to study the pathogenesis of liver disease and to screen and evaluate treatment strategies [36,37]. Each model has its limitations and advantages. For instance, PHHs can preserve the genetic information and metabolic function of their in vivo counterparts. They are suitable for studying toxicity, metabolism, viral infection, and congenital disease. However, their adoption is significantly constrained by a lack of donors and limited proliferation capacity in vitro. 38 On the other hand, immortalized cell lines derived from hepatoblastoma (hepG2) or hepatocellular carcinoma (HCC) (Hep3B) provide a renewable cell source, but they lack multiple hepatocyte functions. Their cancerous features and genomic instability also remain a concern for many applications [39]. During the past decade, hepatic organoids have emerged as a new human in vitro option for modeling multiple liver diseases, including hepatitis, NAFLD, ALD, monogenic disorder, and primary liver cancer (PLC) (Tables 1–4). In general, liver organoids have significantly increased the feasibility of establishing patient-specific models. Additionally, they incorporate multiple liver cell types to model disease pathophysiology and enable the recapitulation of structural changes observed during tissue injury and disease progression. Liver organoids of different cellular compositions, sizes, structures, and functions can be derived from PSCs and human liver tissue (Fig. 1) [40,41]. In this review, we follow the consensus nomenclature of Marsee et al. [42],which reflects the cellular origins of tissues and the cellular composition and function of the organoid. Several reviews have provided in-depth discussions about how various liver organoids (Fig. 1) can be generated using different cell culture systems [40,41]. In this review, we focus on how PSC-derived and tissue-derived liver organoid systems are being used to advance the study of major liver diseases.
- HEREDITARY LIVER DISEASES
- HEREDITARY LIVER DISEASES
Monogenic liver diseases are caused by single-gene mutations. These diseases include conditions that directly induce injury in liver parenchymal cells and diseases in which the liver is intact and the phenotype develops in other parts of the body [43]. The prevalence of individual diseases caused by a single-gene mutation is low, with such diseases occurring in approximately 1% of newborns [43]. Current pharmacological interventions include enzyme replacement therapy and chelation therapy to enhance toxin excretion [44,45]. Unfortunately, those therapies are usually expensive and have poor cost-effectiveness. Gene therapy has emerged as a promising therapeutic option [46]. However, the application of such therapy is limited by the efficacy of the delivery system and the expression of the therapeutic gene. In addition, adverse immune responses and safety concerns about the delivery vectors pose significant difficulties to clinical translation. Liver transplantation remains the standard intervention for many hereditary liver diseases.The successful derivation of organoids from diseased tissue provides a new resource for modeling and understanding hereditary disease and searching for novel therapeutic targets (Table 1). Organoids derived from diseased tissues recapitulate the genomic backgrounds of individual patients and retain the functional dysregulations of monogenetic disorders [28,47-49]. In addition, genome editing technology enables the precise insertion and correction of mutated sequences for further mechanistic study. Clevers’s group reported the first platform for isolating bipotent intrahepatic cholangiocyte organoids (ICOs) from healthy and diseased tissues [47]. Bipotent ICOs can be further differentiated in vitro into functional hepatocytes that secrete albumin, exhibit CYP3A4 activity, and give rise to new hepatocytes upon engraftment in mice. This platform thus makes it possible to model adult liver functions and mimic disease pathophysiology in vitro. Those authors first demonstrated that possibility by isolating diseased organoids from patients with a1-antitrypsin (A1AT) deficiency or Alagille syndrome (ALGS). A1AT deficiency is a genetic disorder caused by the defective production of the A1AT protein in hepatocytes [47]. The A1AT protein is secreted in the lungs to protect the lungs against damage caused by the neutrophil elastase. A1AT deficiency is characterized by an excessive accumulation of abnormal A1AT protein in the liver and reduced A1AT levels in the lung, resulting in the injury to the alveoli [49,50]. Differentiated organoids from A1AT patients displayed normal albumin secretion and low-density lipoprotein (LDL) uptake ability, and the cells acquired the hallmarks of A1AT-deficient patients [47,49]. The patient-derived organoids exhibited reduced A1AT secretion compared with the healthy control organoids, and aggregates of abnormal A1AT increased. These phenotypes are similar to the phenotypes observed in diseased tissue. ALGS is a common monogenic liver disease resulting from a loss-of-function mutation in the JAG1 gene; it affects various organs, particularly the heart, liver, eyes, and vertebrae. The JAG-NOTCH signaling pathway is highly conserved and critical in the development of multiple organs and determination of cellular fates. The clinical manifestation of ALGS in the liver includes bile duct malformation [51]. Patients can present with impaired biliary differentiation, duct paucity, or chronic cholestasis. Isolated patient bipotent organoids are indistinguishable from healthy organoids. However, upon differentiation, the patient organoids failed to upregulate cholangiocyte markers such as cytokeratin 19 and cytokeratin 7 [47]. Subsequently, the organoids lost their morphology and underwent apoptosis. In summary, that pioneering study offered a novel approach for deriving renewable, patient-specific, primary cell models. The rapid adoption of that technique by other research teams for similar genetic liver disease studies supports the robustness of the ICO platform. Gómez‐Mariano’s team isolated diseased ICOs from patients with heterozygous and homozygous mutations in the A1AT-encoding gene SERPINA1, and the organoids with corresponding genotypes exhibited different protein deficiency levels [49]. Those authors further demonstrated that the organoids could respond to external stimuli, such as oncostatin M, which is a well-known inducer of A1AT expression. Other teams have further extended the application of this technique to animal models, including genetically engineered mouse models (GEMMs) [51] and large animal models such as canines (Table 1) [52,53].iPSC technology was a landmark breakthrough that enabled the efficient establishment of patient-specific cell models [54]. Models of genetic liver diseases can be established using iPSCs generated from patients with the disease or via gene editing of healthy iPSC cell lines. Liver organoid models of those diseases can subsequently be established from the iPSCs using various differentiation strategies (Fig. 1). PSC-derived organoids range from simple single-cell-type hepatocyte or cholangiocyte organoids with cystic structures [55-57] to more complex multicellular organoids such as parenchymal hepatobiliary organoids and multicellular liver organoids containing both parenchymal and nonparenchymal cell types [23,58,59]. The structural features present in the organoids allow researchers to recapitulate disease phenotypes in a dish. Harnessing the cystic structure formed by cholangiocyte-like cell (CLC) organoids, Sampazio et al. [56] were able to model the disruption of molecules and fluid transport that occur in cholangiopathy-inducing genetic diseases such as ALGS, polycystic liver disease, and cystic fibrosis (CF). CF is one of the most common autosomal-recessive genetic diseases in the Western population and is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene [60]. The most frequent CF mutation seen in patients is the deletion of phenylalanine 508 (ΔF508), which results in rapid endoplasmic reticulum degradation of misfolded CFTR. The loss of CFTR results in the disruption of chloride transport in cells. CLC organoids derived from the iPSCs of CF patients exhibited disrupted intracellular chloride transport and chloride transport into the organoid lumen. Given the close relationship between chloride transport and fluid secretion in cholangiocytes, those authors further demonstrated that chloride transport dysfunction also affected fluid transport into the organoid lumen. Notably, they demonstrated that the organoids could be used to validate the efficacy of the experimental drug VX809, which is currently in phase III clinical trials. Using a similar strategy, other researchers have reported iPSC-derived liver organoid models of ALGS [61], citrullinemia type 1 [62], and Wolman disease [58]. All those liver organoids exhibited the hallmarks of their respective genetic disease, and subsequent correction with gene-editing approaches reversed the phenotypes [61]. Although many of those proof-of-concept studies demonstrated the potential of generating liver organoids with patient-specific genotypes, how those organoids could advance the study of monogenic liver diseases or replacement therapy remains to be explored. For example, future studies could examine how the different cell types within the liver interact to modulate disease development and progression. In addition, many of the iPSC-derived organoids described herein face the inherent challenge that the hepatocytes generated are immature [63]. These PSC-derived cells are often referred to as hepatocyte-like cells (HLCs) because they closely resemble fetal liver hepatocytes and express markers such as alpha-fetoprotein (AFP) that are absent from adult liver hepatocytes. Of concern, these immature cells might also lack adult hepatocyte functions, such as selected drug-induced cytochrome p450 activity [64]. Significant progress has been achieved in identifying the molecular pathways necessary to induce maturity in fetal hepatocytes, and subsequent optimized culture conditions have enabled considerable progress in PSC-derived HLCs [65]. Therefore, culture conditions for liver organoid models could similarly be optimized in the future to enhance the functional maturity of the cells.
- PRIMARY LIVER CANCER
- PRIMARY LIVER CANCER
- Tumoroid models for PLC
- Tumoroid models for PLC
Large PLC cohort studies across different ethnicities have reported significant interpatient diversity [92,93]. Therefore, building a patient-specific model to support a personalized treatment strategy is crucial for the effective treatment of this disease. The landmark study by Broutier et al. [35] demonstrated the potential of the organoid platform to achieve that breakthrough. That team showed that the ICO culture system could derive tumoroid cultures from surgical tumor resections of HCC, CCC, and cHCC-CCC [36]. Tumoroids isolated from the surgical resections preserved the mutational profile, transcriptome, and histological features of the original tumor, even after long-term (months) culture. Importantly, a proof of concept study further demonstrated the use of those organoids to 1) identify potential prognostic markers by comparing the gene expression profiles of tumoroids to ICOs generated from adjacent nontumor tissue and 2) evaluate the tumor subtype and patient-specific drug response. A parallel effort by Saltsmann et al. [67] further showed that the platform can derive tumoroid models from hepatoblastoma, for which human models have been scant. In another follow-up study, Nuciforo et al. [89] demonstrated that the culture platform could also work with needle biopsy samples and their much smaller tumor tissue volumes. That achievement could be essential for the use of future precision therapeutics, for which tumoroids could serve as patient avatars for in vitro screening to identify the most suitable treatment options [89]. To use the tumoroid platform to capture intratumor heterogeneity, Li et al. [94] divided a tumor into multiple sections and generated tumoroids for each section. In that way, 27 tumoroid lines were obtained from 5 HCC and CCC patients, and the responses to 129 cancer drugs were assessed. The drug response study revealed significant intratumoral functional heterogeneity in the tumoroids derived from different tumor sectors. In addition to human tissue, ICO organoid cultures can be used with GEMMs in mechanistic studies of HCC development. Cao et al. [95] used tumor organoids isolated from reporter mouse models to study the function of LGR5+ cells in tumor initiation. The LGR5+ cells expressed genes associated with multiple hepatic stem cell–related pathways, including the Wnt signaling, Notch signaling, and BMP signaling pathways. Those cells exhibited an increased capacity for tumoroid formation in culture and tumorigenesis in mice and increased resistance to conventional anticancer therapy compared with the controls.In addition to tumoroid cultures, organoids derived from healthy liver tissues can also be used to model the development of liver cancer. Using human ICOs derived from healthy tissues, Artegiani et al. [96] revealed how BRCA1 associated protein-1 (BAP1) potentially functions as a tumor suppressor in ductal cells. Frequent mutations in the BAP1 gene have been reported by exome profiling studies of CCC patients [97]. However, the exact mechanism by which the chromatin modifier affects tumor initiation or progression remains unclear. Those authors demonstrated that BAP1 depletion results in the loss of epithelial cell identity in p53-depleted cholangiocyte organoids. Specifically, the organoid morphology transitioned from translucent cystic spheroids to dense organoids with thickened cell layers and invaginations, resembling tumoroids derived from PLC. BAP1 was found to modulate that phenotypic change via the transcriptional regulation of genes important for junction formation and cytoskeletal components. That study highlighted the immense potential of healthy organoids as an in vitro platform to validate the potential tumor suppressors and oncogenes identified in many PLC cohort studies. Using a similar approach, Yang et al. [98] were able to model YAP1-induced hepatoblastoma development with hepatocyte organoids (HOs) generated from healthy fetal livers. The HOs derived from fetal livers resembled hepatoblasts with high AFP expression. Those authors demonstrated that YAP1 activation is sufficient for oncogenic transformation. The process likely involves epigenetic changes driven by the histone methyltransferase G9a, and inhibitors targeting that molecule were shown to be promising therapeutics for YAP1-driven hepatoblastoma.The abovementioned studies mainly used the tissue-derived organoid culture method pioneered by Hans Clevers and Meritxell Huch [28,47,99]. Taking a different approach, Sun et al. [100] generated HCC tumoroids using the transdifferentiation approach (Fig. 2). Hui’s group previously reported the successful transdifferentiation of fibroblasts to human-induced hepatocytes, and they found that the overexpression of the SV40 large T antigen enabled stable expansion [101]. By overexpressing c-Myc, a frequent HCC driver gene [102,103], those authors induced HCC in organoids that lost their hepatic functions and begin to form tumors when they were engrafted into immunodeficient mice. Using that platform, those authors revealed that Myc-driven HCC development likely involves dysregulated alterations in mitochondria-associated EndoReticulum (ER) membranes and mitochondrial functions. In addition, that team used the system to validate that human hepatocytes can be the cellular origins of CCC. Notably, the RAS-mediated CCC oncogenic transformation was evident only in the 3D organoid cultures, not their 2D counterparts, highlighting the value of modeling human diseases in organoid cultures.Although organoid culture systems have enabled significant breakthroughs in modeling PLCs, several limitations remain. A major hurdle for adopting the ICO culture approach for precision medicine is the low tumoroid derivation efficiency that has been reported. The average success rate of ~30% reported in the abovementioned studies is much lower than the tumoroid culture efficacy achieved in other cancer indications, such as colon and pancreatic cancer [32,33]. Broutier et al. [35] reported that the low success rate is closely associated with the proportion of KI67+ proliferative cells in the biopsy. However, Nuciforo et al. [89] reported that tumoroids could only be derived from moderately and poorly differentiated tumors. Those results suggest that the culture of liver cancer organoids is limited to a proliferative and aggressive subset of cancers and might fail to capture the entire disease spectrum. Therefore the loss of cancer cell heterogeneity in the organoid culture may affect the in vitro prediction of clinical drug responses. Another concern is the presence of healthy organoids in tumoroid cultures. Essential growth factors should be removed from the culture in the first two weeks, and some of the healthy organoids must be mechanically removed from the tumoroid culture to reduce contamination [35]. However, long-term, stable tumoroid expansion still depends on using a complete growth factor medium that supports the culture of healthy organoids. This outgrowth of noncancer, healthy-organoid contaminants has also occurred in other tumoroid cultures that have used similar tissue-derived organoid culture approaches in lung [104], pancreas [33], and prostate [105] cancer research. Although treating tumoroid cultures with p53 pathway activators such as Nutlin3 can induce cell death in noncancer organoids, that strategy might not work for all cancer subtypes due to interpatient and intratumor heterogeneity. The future optimization of culture conditions will be critical to enabling the clinical use of precision medicine. A related issue in tumoroid culture is the clonal selection that occurs during long-term passaging. Although organoid culture potentially supports multiple cancer cell clones during derivation and early maintenance, that diversity is likely lost over time due to competitive growth between cancer clones. That process was reportedly observed in the Colorectal cancer (CRC) organoid biobank established by Fuji et al. [106]. Those authors co-cultured three pairs of CRC tumoroids, each pair derived from different regions of the same tumor. After 4–6 weeks, all three paired cultures were dominated by a single population of tumoroids. Although that loss of cancer cell heterogeneity was not investigated in the abovementioned PLC tumoroid studies, this issue is likely to be pervasive across tumoroid cultures for various cancer indications.
PLCs are some of the most malignant tumors and the fourth-leading cause of cancer-related death worldwide [66]. The types of liver cancer include HCC, which accounts for the majority (70–85%) of PLCs, cholangiocarcinoma (CCC), and combined hepatocellular cholangiocarcinoma (cHCC-CCC), which exhibits features of both HCC and CCC [66]. Hepatoblastoma is a rare liver tumor that predominantly originates in early childhood; it is the most common pediatric liver malignancy [66,67]. The main etiologies of PLC include hepatitis virus infections, alcohol abuse, NAFLD, and aflatoxins [66,68]. With the development of vaccinations and antiviral treatments, the major cause for PLC has gradually shifted from viral infections to NASH, in correlation with the growing global obesity pandemic [69,70].Conventional treatment options for liver cancer patients include surgical resection, radiofrequency ablation, liver transplantation and multiple-kinase inhibitors such as, Sorafenib and Lenvatinib [71]. Unfortunately, the median progression-free survival time remains less than six months for most of these treatments [72]. With the emerging success of immunotherapy in targeting advanced-stage lung cancer and melanoma, immune checkpoint inhibitors have also been explored to treat liver cancer [73]. Currently, the combination of antibodies blocking PD-L1 and VEGF is the 1st line of treatment for HCC patients with unresectable tumours. However, the available data from clinical trials showed that less than half of the patients responded to the treatment [74].Liver cancer manifestation is highly complex and involves the dysregulation of multiple genes and signaling pathways as the disease progresses [75]. Comprehensive genome and transcriptome profiling studies have been conducted to reveal the genetic landscapes of cancer cells and unravel the mechanisms of HCC initiation and progression [76-78]. Mutations in genes such as TP53, CTNNB1, CDKN2A, and TERT have been commonly identified in different patient cohorts [78-80]. Various underlying diseases and etiologies can induce tumorigenesis in the liver. Postinfection, the HBV genome potentially integrates with host cells and activates genes relevant to the cell cycle and proliferation such as TERT [81-83], stabilizing the telomeric ends of chromosomes to support unlimited cell expansion. Diseases such as NAFLD and long-term drug and alcohol abuse induce chronic inflammation in the hepatic niche [84]. Constant exposure to inflammatory cytokines promotes genetic insults that lead to unique gene mutations and epigenetic profile changes that favor cell survival under harsh conditions [85,86]. The interplay among those different disease etiologies creates an environment that promotes oncogenic transformation, clonal evolution, and the expansion of diverse cancer-initiating cells. Thus, liver cancers, such as HCC, are reported to exhibit significant interpatient and intratumoral heterogeneity.For decades, researchers have derived 2D monolayer cancer cell lines from tumors and created mouse models to understand the disease and search for therapeutic targets. These immortalized cell lines are highly expandable at low cost, enabling high-throughput drug screening applications. However, a single immortalized cancer cell line only partially captures the tumor profile and constantly acquires new mutations during culture [8,40]. The lack of intratumor heterogeneity produces an inaccurate reflection of patient responses when such cell lines are used for drug screening. Moreover, a monolayer cell culture limits cell–cell interactions to a horizontal plane, and the nutrient and oxygen gradients observed in tumors are absent [8]. A study by Takai et al. [87] demonstrated that immortalized HCC cell lines cultured in 3D produce structural features resembling the glandular epithelium observed in vivo, and exhibit they increased resistance to chemotherapeutic agents. The cells in 3D also showed increased sensitivity to the TGF/β-induced epithelial–mesenchymal transition. Correspondingly, after engraftment in mice, the cells from the spheroids formed tumors more efficiently that metastasized more frequently than the cells cultured in 2D. Such studies highlight the advantages of alternative 3D in vitro models of PLCs. The advent of organoid culture thus presents a long-sought alternative strategy for creating human PLC models (Fig. 2). Commonly used immortalized cell lines have been derived and grown with undefined serum-supported media in monolayer cultures [88]. Although such serum-based culture conditions have enabled the creation of widely used immortalized cell lines, they fail to recapitulate the interpatient and intratumor heterogeneity observed in HCC tumors [89]. In contrast, organoid culture platforms use a chemically defined nutrient- and growth factor (cytokines and small molecules)–enriched medium that potentially supports the growth of various cancer cell clones. Another critical advantage of the organoid platform is the ability to culture both cancerous and healthy tissue from the same patient in parallel, which creates opportunities to study tumor development without interfering with individual genetic backgrounds (Fig. 2) [35]. The liver cancer tumoroid platform has contributed significantly toward understanding cancer cell biology, and it opens new possibilities for precision treatment in the clinic (Table 2) [8,90,91].
- VIRUS-DRIVEN LIVER DISEASES
- VIRUS-DRIVEN LIVER DISEASES
- Liver organoids for viral-hepatitis modeling
- Liver organoids for viral-hepatitis modeling
HLCs derived from PSCs are one source of renewable primary cells for modeling HBV infection [127-129], and multicellular organoids derived from iPSCs have also been explored (Table 3). Nie et al. [130] generated functional liver organoids (LOs) using the differentiation of PSC-derived endoderm cells co-cultured with mesenchymal stem cells and human umbilical vein endothelial cells. In that study, they demonstrated the advantages of modeling HBV with LOs, compared with HLCs generated from the same iPSCs. The LOs were shown to be more functional than their HLC counterparts, as reflected by the increased expression of cytochrome P450 genes, albumin and urea production, and CYP3A4 activity. Notably, although NTCP expression in the HLCs and LOs was comparable, HBV infection was much more efficient in the LOs, as reflected by the increased levels of pgRNA, intracellular viral DNA, and cccDNA. Correspondingly, a higher level of virion release was also detected in the LOs. Intriguingly, those authors discovered that LOs generated from different patient iPSCs demonstrated varying levels of HBV infectivity. Those results further support the role of host-specific factors in modulating HBV infection, which could account for variations observed within specific ethnic populations [130]. In addition, those authors demonstrated that the LOs could maintain their function in culture for up to 15 days longer than HLCs, enabling the evaluation of extended HBV infection. HBV-infected hepatocytes in the LOs generated virions that could further infect primary hepatocytes. Furthermore, the hepatocytes exhibited cytopathic responses, such as loss of hepatic function, injury marker expression, and structural changes, including increased cytoplasmic vacuoles and the loss of membrane microvilli. Although the existence of other cell types seemingly improves the hepatic functions of the LOs, it remains to be investigated whether the other cell types play a direct role in modulating viral infection. Further HBV modeling studies with multicellular organoids (Fig. 1) with different cellular compositions and structural features will advance knowledge about the roles of other liver cell types in HBV infection and progression.The spectrum of patient profiles observed in HBV is diverse. Patient disease status ranges from individuals who have a high viral load in their blood but no hepatitis symptoms to individuals with hepatitis symptoms and a low viral load [110]. Such disparate patient responses indicate the need to develop patient-specific HBV models that address host factor–specific responses (Fig. 3). To that end, De Crignis et al. [131] used the ICO platform to successfully generate HBV organoid models derived from healthy and HBV-infected individuals. The organoid derivation efficiency was comparable from healthy and diseased tissues. The healthy organoids (HOs) and disease organoids (DOs) also exhibited similar proliferation and differentiation capacities. HOs infected with the HBV virus exhibited the hallmarks of infection, including cccDNA formation and viral antigen expression. Of note, the organoids further exhibited dose-dependent toxicity in response to NRTIs, which was not observed in HepG2 cells. This further supports the potential application of HBV patient–derived ICOs for testing idiopathic drug responses and toxicity. Those authors also demonstrated the amenability of the ICOs to genetic modification by creating transgenic cell lines that overexpressed NTCP and HBV virus–producing cell lines. Intriguingly, the NTCP-overexpressing ICOs cultured in expansion medium (nondifferentiated) did not exhibit greater HBV susceptibility than the differentiated ICOs. This observation further validates that hepatocytes need other factors to be susceptible to HBV infection, and it could account for the absence of detectable virus in ICOs derived from HBV patients. Nonetheless, those authors reported the detection of HBx coding region integration in 5 out of the 6 organoids assayed. In addition to viral replication, the HBx antigen has been implicated in immune protection from host cells and HCC development [132]. Thus, those organoids will likely be helpful in gaining mechanistic insights into how the antigen functions and promotes those activities. Those authors also identified a set of genes that is enriched in DOs, compared with HOs. The DO gene signature is reflected in HBV and HCC patient tissues, so it could potentially serve as an early prognostic marker for HCC development.Although HEV infection is rare compared with HBV and HCV infection, the virus is known to infect pregnant people and have severe effects on maternal and fetal outcomes in resource-limited regions [133]. Unfortunately, available therapeutic options, such as antiviral ribavirin and pegylated interferon-α, are contraindicated during pregnancy [134]. Adopting the ICO organoid culture system, Li et al. [135] established fetal and adult liver organoid models of HEV infection to investigate potential therapeutic options. They demonstrated the feasibility of establishing HEV infection in the organoids and validated the broad virus tropism in cholangiocytes and hepatocytes and virus release in the apical surfaces of both cell types. In addition, they performed a small-scale drug screen with the organoids and demonstrated the potential of the antiviral homoharringtonine for HEV therapy. By manipulating the HEV variant introduced into the organoids, they further highlighted the utility of homoharringtonine in treating the ribavirin-resistant p6G1634R HEV variant [135].- Other virus-driven liver disease studies with organoid models
- Other virus-driven liver disease studies with organoid models
The major symptoms of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) virus infection are associated with the respiratory system. However, further clinical evidence and virus tropism studies suggest that the SARS-CoV-2 virus might infect cells in other organs, including the liver [136,141,142]. Importantly, a significant number of SARS-CoV-2 patients with severe symptoms were also reported to have abnormal liver enzyme levels [142,143]. To further understand how the liver could be affected by the SARS-CoV-2 virus, multiple groups have used the ICO culture [138,140] and PSC-derived organoid culture methods [136,137] to validate direct viral infection in hepatocytes and cholangiocytes, as well as to investigate disease manifestations in individual cells (Table 3). The discovery of ACE2 and TMPRSS as the proteins that mediate SARS-CoV-2 tropism prompted scientists to identify other potential cell types that the virus could infect by using singlecell tissue profiling experiments [144]. Analyzing single-cell profiles of liver tissue to identify SARS-CoV-2 target cell types has been challenging due to the low levels of ACE2 expression detected on a single-cell sequencing platform and the low percentage of cells expressing those proteins. Liver organoids generated from PSCs or tissue have helped to validate that the virus can infect both parenchymal cell types. Of note, a study by McCarron et al. [140] highlighted that NASH patient–derived organoids were particularly susceptible to SARS-CoV-2 infection due to the increased expression of ubiquitin D, which is an inhibitor of RNA virus–induced interferon signaling. This interesting finding indicates that direct virus tropism could contribute to the increase in abnormal liver functions and longer viral shedding time observed in NAFLD patients [145].In summary, organoid platforms have been used to establish human models for various virus-driven liver diseases. These models have produced mechanistic insights about viral tropism and enabled the derivation of patient-specific models to address idiosyncratic responses to viral infections. The future growth of a biobank of hepatitis patient–derived organoids would likely accelerate the identification of hostspecific factors that modulate viral tropism and life cycle (Fig. 3).
The human liver is a highly vascularized organ that is exposed to many external pathogens in the blood [1]. Common pathogens that infect hepatocytes include malaria-causing Plasmodium parasites and the hepatitis virus [108]. In contrast to Plasmodium parasites, the hepatitis virus affects host cell function and integrity, inducing liver inflammation and injury. Chronic hepatitis can progress to end-stage liver cirrhosis or liver cancer, and viral hepatitis was estimated to cause 1.34 million deaths globally and HBV and HCV infections accounted for 96% of mortality reported [109].Understanding how these viral proteins and host–virus protein interactions function in each essential step of the viral life cycle has enabled the discovery of therapeutics. In HBV therapy, nucleoside reverse transcriptase inhibitors (NRTIs), which target HBV polymerase to disrupt viral genome replication, are currently prescribed to suppress viral levels in the liver [110,111]. Drugs targeting the interaction between L-HBs and NTCP, such as the myristoylated peptide Myrcludex B, have also been shown to reduce viral infection in preclinical models and early clinical trials [112]. Although targeting many of those viral pathways has enabled the effective management of the disease in patients with few liver complications, a cure is still unavailable because viral covalently closed circular DNA (cccDNA) persists in the host cell as a mini-chromosome or is integrated into the host genome [113]. In stark comparison, HCV is now widely considered to be a curable disease thanks to a range of direct-acting antiviral (DAA) drugs that target various viral nonstructural proteins [114]. Combinations of DAA drugs now enable 80–95% of HCV patients to be cured.In a previous review of HCV research breakthroughs that enabled the rapid development of therapies, in vitro modeling for HCV replication was demonstrated to play a crucial role [114]. The discovery that the overexpression of cDNA encoding various viral genotypes induces full viral replication in hepatic cell lines enabled the rapid investigation and screening of therapeutics targeting different parts of the viral life cycle [115,115]. In vitro models continue to offer researchers mechanistic insights into how the HCV virus infects the host [117]. Human hepatic cell models, each with advantages and limitations, have also been used for HBV infection studies. Immortalized cancer cell lines, such as HepG2 and Huh7, are easy to culture and proliferate rapidly. However, they do not support HBV infection due to a lack of endogenous NTCP expression, so exogenous expression of NTCP is used in those cell lines to solve that problem. Although the ectopic expression of NTCP in those cells has proved useful [118-120], immortalized and mutating cancer cells might have lost cellular components that regulate viral entry. In light of that concern, HepaRG, a tumor-derived hepatic progenitor cell line capable of generating NTCP-expressing HLCs, is increasingly being adopted for HBV modeling [121-125]. However, the limitations of HepaRG in HBV modeling include low infection efficiency (requiring a high infection dose of >1,000 GE/cell) and low levels of cccDNA [126].
- NONALCOHOLIC FATTY LIVER DISEASE
- NONALCOHOLIC FATTY LIVER DISEASE
- Modeling NAFLD with human liver organoids
- Modeling NAFLD with human liver organoids
Although a fatty liver begins with the excessive accumulation of lipids in hepatocytes, disease progression involves multiple NPCs such as KCs and HSCs [171]. Those cells are central to the chronic inflammatory response and fibrogenesis that account for the hepatocyte cell loss and remodeling of the tissue architecture that occur as NAFLD progresses to NASH. The observed progression rate for NAFLD to NASH is ~14 years, and the progression rate in NASH patients is ~7 years per fibrosis stage [183]. This long time period highlights the complex interplay among different cell types that mediates NAFLD progression and the need to develop more advanced multicellular models. Ouchi et al. [58] reported one of the first PSC-derived multicellular organoids containing most liver parenchymal and NPC types, including HLCs, KCs, and HSCs. Although the cells in the organoids lacked a structural organization resembling that in tissue, the presence of cells resembling all the different liver cell types enabled the recapitulation of many NAFLD phenotypes upon treatment with FFAs. The triglyceride levels increased in the FFA-treated organoids, and the hepatocytes accumulated large lipid droplets and swelled. In addition, inflammatory responses were observed via the detection of cytokines such as TNF-α and IL-8. Furthermore, the FFA-treated organoids exhibited the hallmarks of fibrogenesis, including elevated P3NP levels, an increased number of α-smooth muscle actin–positive HSCs, and increased collagen deposition. Those authors also demonstrated that fibrogenesis in the organoids correlated with increased organoid stiffness, as measured by atomic force microscopy. Overall, that pilot study demonstrated the utility of multicellular organoids in modeling different phenotypes of NAFLD.To capture the structural features of liver tissue, Ramli et al. used a stepwise PSC differentiation approach to generate hepatobiliary organoids with a functional bile canaliculi network [59]. Using a cocktail of palmitic and oleic acids, those authors also demonstrated the feasibility of generating NAFLD organoids in vitro that exhibit steatosis features and express transcriptome profiles resembling those of NAFLD patient tissues. The development of imaging techniques and analysis has revealed structural changes in the bile canaliculi during NAFLD progression [184]. Specifically, Segovia-Miranda et al. [184] reported that the bile canaliculi begin to narrow and shorten in NAFLD patients before NASH develops. Remarkably, the bile canaliculi network in the hepatobiliary organoid treated with FFA cocktails demonstrated a similar response. Future studies using these organoids will help provide mechanistic insights into those structural changes. Interestingly, those authors detected the proliferation of cholangiocyte cyst structures in the FFA-treated hepatobiliary organoids, in parallel with an increasing proportion of dying hepatocytes. This observation is similar to the ductular reaction (DR) observed in NASH patient tissues. However, the lack of NPCs in the organoids prevented further investigation of the DR-induced fibrogenesis observed in animal models [185]. Overall, that study demonstrated the potential of organoids to recapitulate structural changes observed in vivo.The organoids described thus far are small (less than a millimeter) and not compatible with modeling larger structural features such as the liver lobules and vasculature. Hepatocytes in the liver lobules are distributed across zones with different levels of nutrients and oxygen [186]. It would be interesting to recapitulate those structural features to understand how liver diseases, including NAFLD, manifest differently across each lobule. To create a sizable physiological human liver model, Collin de l’Hortet et al. [180] used a co-culture system of iPSC-derived hepatic cells and NPCs in a decellularized rat liver scaffold. By using a peristaltic pump to introduce cells into the large tissue scaffold and continuously provide nutrients to all parts of the tissue-like culture, the group maintained the largest (centimeter-size) in vitro liver organoid to date. Notably, the vascular flow system was essential to achieve an NAFLD phenotype across the entire organoid. Compared with previous organoids, in which each NAFLD phenotype was assayed using a different molecular assay and multiple organoids, the achievement of a tissue-like organoid culture allowed those authors to perform histological NAFLD activity score (NAS) scoring, similar to the test used with tissue sections from patients. One key advantage of such a co-culture system, compared with the abovementioned stepwise differentiation approach, is the potential use of transgenic cells to study gene function in the cell type of interest. SIRT1 expression is downregulated in the hepatocytes of NASH patients, and SIRT1 knockout in mouse livers induces NAFLD [187]. Using SIRT1-knockout iPSC cell lines to generate hepatocytes for organoid formation, Collin de l’Hortet et al. [180] achieved the specific depletion of SIRT1 in those hepatocytes. They thus revealed the role of SIRT1 in regulating lipolysis and de novo lipogenesis in hepatocytes, and they demonstrated that SIRT1 depletion induced an NAFLD phenotype in the organoid. In follow-up studies, it would be interesting to explore whether that system can recapitulate hepatocyte zonation across large organoids and be further refined to achieve bidirectional sinusoidal and bile canaliculi flow. In particular, that study highlights the value of integrating bioengineering techniques to generate advanced liver organoid cultures. An organ-on-a-chip culture that uses microfluidics techniques is also frequently harnessed to create 3D liver models [188]. Microfluidics platforms enable the precision control of media flow and cellular placement to create cellular interactions and spatial organizations that resemble the liver architecture. Furthermore, integrating nanosensors and electronics in the chip enables researchers to monitor cellular changes during disease modeling. We expect the increasing use of such microfluidics platforms for organoid cultures to generate more complex 3D liver models for various applications [189].The high failure rate observed in NASH clinical trials remains a crucial factor in the lack of approved therapeutics for NASH patients [190]. Challenges for drug development include the complex interactions of multiple disease drivers and the need to correct metabolic dysregulation and reverse fibrotic scars, which are likely driven by different mechanisms. Another factor that probably contributes to the high failure rate, as well as future challenges in prescribing an approved drug, is the influence of patient genotype on drug efficacy. That became evident in the early phase 2 trials of obeticholic acid. This drug was proved effective in a Western population but failed in a trial with a Japanese population [191,192]. Therefore, patient-specific models are needed to further identify the patient genotypes that modulate NASH development and evaluate patient-specific responses to NASH drugs. The further accumulation of these patient models into a biobank of comprehensive clinical, molecular, and phenotype profiles will enable the future correlation of patient genotypes and phenotype responses for pharmacogenomic applications (Fig. 3). Both iPSC-derived and tissue-derived organoid systems are potentially suitable for this application.In a proof of concept study, Kimura et al. [181] generated NAFLD organoid models from 24 patient-specific iPSC cell lines to identify patient-specific genotypes that correlate with NAFLD phenotype development. They established a high-throughput en masse differentiation platform that they termed the Population organoid platform to enable the parallel culture of 24 patient organoids in a single Matrigel droplet and the induction of an NAFLD phenotype. Using a combination of imaging and sequencing readouts, those authors mapped NAFLD phenotype development in the organoids to specific patient genotypes. They demonstrated the utility of their platform by validating the contribution of the GCKRrs1260326:C>T SNP to hepatocyte lipid accumulation via mitochondrial dysregulation. In addition, a retrospective evaluation of clinical studies revealed that that variant contributed to a higher risk of NASH development only in diabetic patients. Furthermore, with the available patient models for the targeted genotype, they were able to identify drugs that could reverse the mitochondrial dysregulation induced by the GCKR variant. Overall, this study lays a strong foundation for the future pharmacogenomic evaluation of NASH therapeutic drugs. This evaluation will likely aid in the identification of patients with genotypes that are suitable for specific drug trials or the exclusion of individuals at high risk for adverse drug responses. Such achievements will significantly accelerate the approval of new drugs for NASH therapy. In a parallel study, McCarron et al. [140] successfully generated long-term expanded ICO organoids from tissue obtained from multiple NASH patients. The organoids from NASH patients exhibited significant upregulation of pro-inflammatory and cytochrome p450–related pathways and liver fibrosis markers. Notably, the upregulation of different markers is idiopathic and not observed across the board. This achievement supports the potential use of tissue-derived organoids for setting up population-specific patient organoid biobanks for precision medicine studies.
NAFLD is a CLD with a high clinical incidence, and it is closely associated with other metabolic disorders such as obesity, diabetes mellitus, and metabolic syndrome [146,147]. Chronic inflammation and hepatic injury promote the progression of NAFLD to NASH and eventually cirrhosis and HCC [148]. The global prevalence of NAFLD in 2018 was ~24.4% [149], and correspondingly, the number of patients with end-stage cirrhosis and HCC due to NASH continues to rise [147]. Although NAFLD patients are clinically asymptomatic, and the condition is relatively benign, NASH is rapidly becoming the leading cause of end-stage liver disease and liver transplantation [150,151]. The high incidence of NAFLD is primarily attributed to diet and lifestyle changes. Irregular lifestyles, such as insufficient sleep and exercise, and the long-term intake of foods high in sugar and fats contribute to excessive fat accumulation in the liver [152]. Common metabolic diseases such as hypertension, hyperglycemia, obesity, and metabolic syndrome are risk factors for NAFLD and NASH development [153-155]. In parallel, genetics also likely play a role in NAFLD development. Familial studies have shown that children of parents with a high liver-fat content are more likely than others to develop NAFLD and have an increased risk of progression to cirrhosis [156]. Genome-wide association studies in multiple NAFLD cohorts have identified enriched single-nucleotide polymorphisms (SNPs) in various genes in NAFLD patients, including apolipoprotein C3, glucokinase regulator (GCKR), membrane-bound O-acyltransferase domain-containing 7, patatin-like phospholipase domain containing 3 (PNPLA3), and transmembrane 6 superfamily member 2 [157]. Furthermore, a protective allele that reduces the risk of NAFLD progression to NASH has been reported in the HSD17B13 gene [158].NAFLD is a continuum of disease ranging from benign hepatic steatosis to NASH and cirrhosis [159], with 43–44% of patients with steatosis progressing to NASH, and 7–30% of patients with NASH progressing to cirrhosis [160,161]. The mechanisms underlying NAFLD progression have yet to be fully elucidated. A two-hit theory was first proposed for the pathogenesis of NAFLD [162]. The first hit involves lipid metabolism disorders and manifests as hepatic steatosis. That results in insulin resistance and leads to increased serum levels of free fatty acids (FFAs) and sensitizes the hepatocytes for the second hit, which comprises endoplasmic reticulum stress, oxidative stress, and mitochondrial dysfunction. Together, the two hits result in hepatocyte damage and the secretion of large amounts of proinflammatory factors that recruit and activate immune cells into a chronic inflammatory response [163]. Accumulating evidence has shifted that traditional two-hit model into a model of multiple, parallel pathogenic influences in which fatty acids, insulin resistance, inflammation, oxidative stress, mitochondrial dysfunction, lipid peroxidation, and endoplasmic reticulum stress combine to induce the onset and development of NAFLD [164]. In addition, gut microbes have been reported to be involved in the development of NAFLD via the regulation of bile acid metabolism, lipid handling, and endotoxin and metabolite production in the gut [165,166]. Chronic inflammation is central to NAFLD disease progression and involves both residential and circulating immune cells, including macrophages and lymphocytes [167,168]. Interactions among the different cell types also involve a slew of inflammatory chemokines and cytokines, such as chemokine ligand 2, tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), IL-1β, IL-17, and IL-18 [169-172].Models of NAFLD have been critical for elucidating the mechanisms underlying this disease and identifying potential therapeutic targets. Multiple diet–induced models and GEMMs have been developed for NAFLD, and most of them recapitulate the histological features of NAFLD [173]. However, only a few studies have captured the metabolic syndromes commonly observed in patients [174], and there have been discrepancies in the NAFLD phenotypes observed in mouse and human models. In follow-up studies about PNPLA3 (the most widely reported NAFLD-associated gene), PNPLA3 depletion in mice did not affect hepatocyte lipid accumulation, which is in contrast to the findings in human in vitro models [175,176]. Human PNPLA3 levels are also much higher in liver tissue than adipose tissue, which is in contrast to rodent PNPLA3, which is more highly expressed in adipose tissue [177,178]. Given the increasing prevalence and lack of therapeutic options for NAFLD, many human models of NAFLD have been created using different hepatic cell lines and 2D and 3D culture platforms, including sophisticated microfluidics platforms [179] and PSC and tissue-derived organoid models (Table 4).
- ORGANOID MODELS FOR DRUG-INDUCED LIVER INJURY AND ALCOHOL-RELATED LIVER DISEASE
- ORGANOID MODELS FOR DRUG-INDUCED LIVER INJURY AND ALCOHOL-RELATED LIVER DISEASE
DILI is a common cause of liver injury and a significant challenge for drug development. Statistically, nearly one-third of drug candidates are withdrawn from the clinic due to adverse drug reactions [193]. Therefore, preclinical drug safety studies are critical to drug development. Drug toxicology studies rely heavily on animal testing in Investigational New Drug applications. However, animal model results have limitations due to significant species-specific physiological differences and the long latency period of some drug-induced injuries [194]. PHHs remain the preferred human in vitro model for DILI testing. The methods used to assess DILI with PHHs are currently limited to the measurement of hepatic enzyme activity, cell structural changes, and cell redox status [195,196]. Therefore, more sophisticated liver models are needed to allow researchers to test for changes in human liver tissue features. Shinozawa et al. [57] reported an iPSC-derived hepatocyte organoid model for high-throughput toxicity screening. The hepatocytes in the cyst-like organoid exhibited polarity and unidirectional transport of bile into the lumen. Using those organoids, those authors set up a screening platform that allows the real-time imaging of bile transport and parallel assays for cell viability. They validated 206 clinically available drugs with reported DILI and showed a high predictive value of ~90% sensitivity and specificity. By treating the organoids with FFA, those authors also demonstrated that the system could be used to investigate DILI under various liver disease backgrounds. In the abovementioned hepatobiliary organoid generated by Ramli et al. [59], the authors demonstrated how it could be used to model cholestatic events in DILI. They performed live-imaging of the organoids with 5(6)-carboxyfluorescein diacetate (CDFDA), which is a fluorescent probe that is transported in the bile system. Treatment of the organoids with the cholestasis-inducing troglitazone resulted in a gradual loss of signals in the bile canaliculi network and the accumulation of CDF (de-esterified CDFDA) in the enlarged cells. Overall, both studies highlight how researchers can harness the structural features observed in organoids to model DILI. We expect that toxicological screening applications will increase in the future with the generation of organoid models that can recapitulate more complex structural features of the liver.In addition to DILI, alcohol abuse is a common cause of liver morbidity and mortality. Globally, approximately 3 million people die each year from alcohol consumption, and 8–20% of heavy drinkers develop cirrhosis [197]. Similar to NAFLD, ALD phenotypes include steatosis, fibrosis, and cirrhosis [223]. A study by Wang et al. [182] explored the use of PSC-derived hepatic organoids to recapitulate the pathogenesis of ALD. They first derived an expandable hepatic progenitor organoid that the enabled long-term expansion and generation of functional cholangiocyte or hepatocyte organoids. Subsequently, they achieved a co-culture of fetal mesenchymal cells and hepatocyte organoids. The organoid expressed ethanol (EtOH) metabolism–associated enzymes such as alcohol dehydrogenase and CYP2E1. When they were treated with EtOH, the cells exhibited increased cellular stress, steatosis, and a proinflammatory response. In addition, the authors detected elevated expression levels of fibrogenesis-related proteins, such as alpha-smooth muscle actin, type 1 collagen, and desmin. In summary, PSC-derived hepatic organoids can also be used to model ALD, but the utility of this model for mechanistic studies and the identification of novel therapeutics remains to be explored.
- CONCLUSION
- CONCLUSION
Liver organoids have provided researchers with an alternative human in vitro model that harbors features of in vivo tissue. Although these models encompass more physiological characteristics of the human organ than monolayer hepatocyte models, organoids should not be considered as a replacement for animal models, at least not in their current state. Organoids still lack many structural features of the tissue, a functional vascular system, and circulating hemopoietic cells. Future improvements to cell culture reagents and tools and the integration of bioengineering platforms will likely bring significant breakthroughs. Nonetheless, the current organoid culture platforms have already produced several advances in modeling human liver diseases. In particular, the ICO platform has enabled the generation of patient-specific liver organoid models for all the conditions discussed. This breakthrough will allow the generation of patient-specific models for various applications, from evaluating host susceptibility to viral infection to liver regenerative therapy (Fig. 3). In the next decade, we expect to see the establishment of sizeable organoid biobanks for various liver diseases. These biobanks will greatly facilitate pharmacogenetic studies by correlating patient genotypes to disease phenotypes and patient drug responses (Fig. 3), laying the foundations for precision therapeutics for treating liver disease.
- ACKNOWLEDGMENTS
- ACKNOWLEDGMENTS
Figures 1 to 3 were created with BioRender.com.
- FOOTNOTES
- FOOTNOTES
-
Authors’ contribution Y. Liu, J.Y Sheng, C.F. Yang and Y.S Chan reviewed articles and wrote the manuscript. Y.S Chan and J.J Ding oversee the review conception and writing.
Conflicts of Interest The authors have no conflicts to disclose.
Figure 1.

Figure 2.
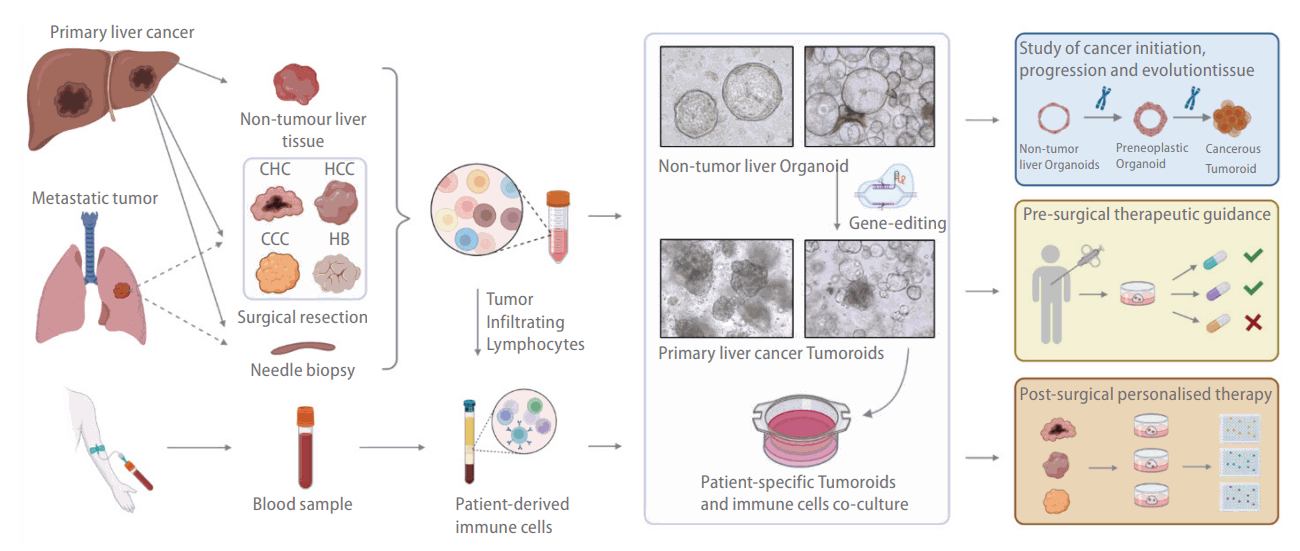
Figure 3.
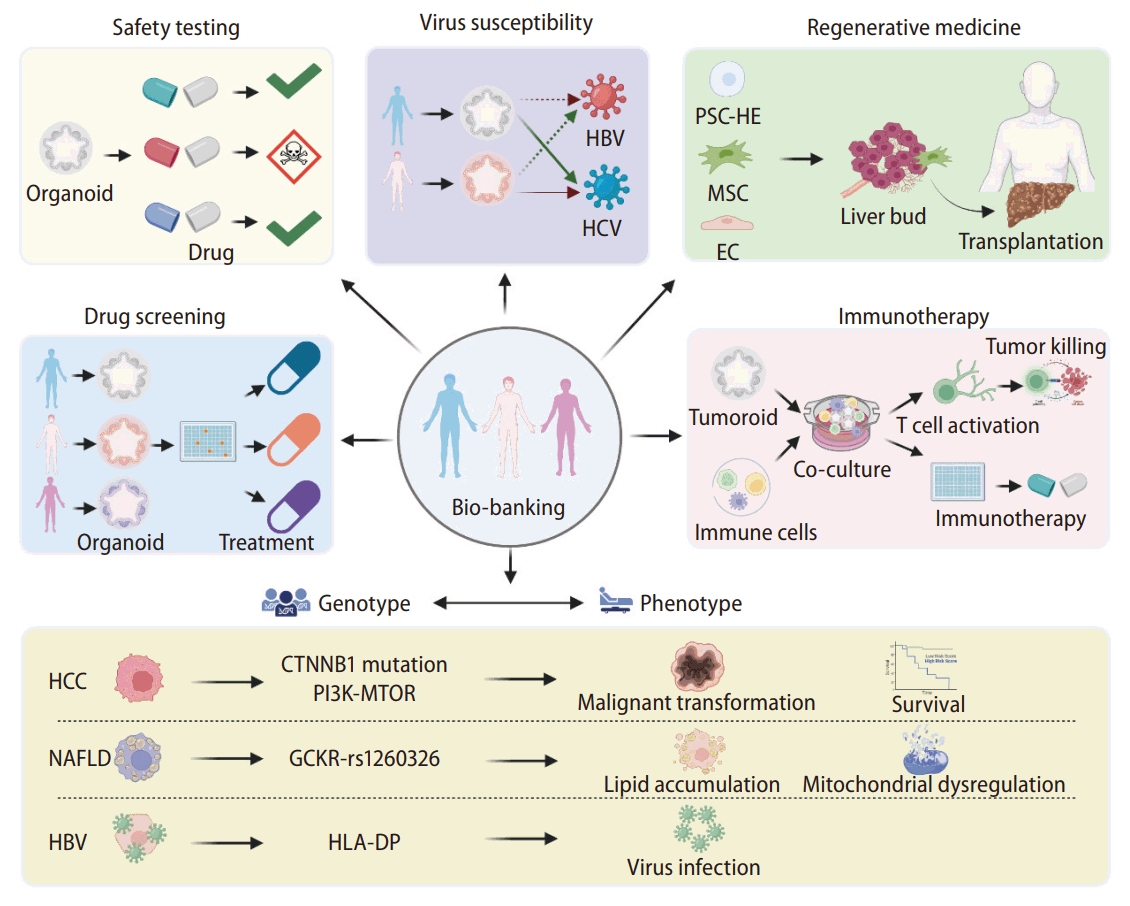
Table 1.
| Reference | Genetic disease | Culture approach | Study highlight |
|---|---|---|---|
| Huch et al. [47], 2015 | Alagille syndrome (ALGS) | ICO culture from liver biopsy | ICOs from AGS patients resemble healthy counterparts in the undifferentiated state but fail to upregulate biliary markers when differentiated to cholangiocytes. This observation corroborates with the reduced bile duct network observed in the patient’s liver. |
| Alpha-1 antitrypsin (A1AT) deficiency | ICOs can also be stably propagated from tissue biopsies from A1AT deficiency patients. A1AT protein aggregates in the hepatocytes of differentiated organoids, and secreted protein is significantly reduced. | ||
| Andersson et al. [51], 2018 | ALGS | ICO culture from GEMM liver biopsy | Organoids from GEMMs exhibit delayed biliary differentiation and morphogenesis and altered structural stability compared to organoids generated from wild-type mice. |
| Guan et al. [61], 2017 | ALGS | Patient-derived iPSC | The patient’s iPSC-derived organoids can recapitulate ALGS liver pathology, including the lack of duct-like structures, diminished biliary transport, and impaired regenerative ability. |
| PSC-derived hepatic organoid culture | The correction of mutation by gene editing rescues organoid functions. | ||
| Gómez-Mariano et al. [49], 2020 | A1AT deficiency | ICO culture from liver biopsy | ICOs were generated from patients with heterozygous and homozygous Z (Glu342Lys) mutation on SERPINA1. |
| Organoids exhibit disease phenotypes, including intracellular aggregation and lower secretion of AAT protein. The patient organoids also showed a differential response to Oncostatin-induced SERPINA1 expression. | |||
| Akbari et al. [62], 2019 | Citrullinemia type 1 | Patient-derived iPSC | Using the ICO culture method, authors derived an expandable Epcam-expressing hepatic organoid (eHEPO) from iPSCs. |
| PSC-derived epithelial organoid culture | Differentiated eHEPOs from citrullinemia patients had a higher accumulation of ammonia and lower ureagenesis levels than eHEPOs from healthy individuals. The phenotypes were reversed with the overexpression of wild-type protein. | ||
| Nantasanti et al. [53], 2015 | Wilson’s disease | Canine liver tissue biopsy | Canine liver-derived organoids exhibit similar morphology, molecular characteristics, and bipotent capacity as human ICOs. |
| Kruitwagen et al. [52], 2020 | ICO culture | Canine Organoids with COMMD1 knockout recapitulated disease phenotypes of intracellular copper accumulation similar to Wilson disease in humans. Genetic restoration of COMMD1 expression in organoid rescues phenotype. Cells can be further engrafted into the canine liver and host survive up to two years after transplantation. | |
| Ouchi et al. [58], 2019 | Wolman disease | PSC-derived multi-tissue organoid | PSC-derived liver organoids containing both parenchymal and NPC types. |
| Organoids generated from a Wolman disease patient’s iPSC exhibit pathological features observed in the liver tissue, including increased lipid accumulation and fibrosis compared to healthy controls. FGF19 treatment ameliorates disease phenotypes in the organoid. |
Table 2.
| Reference | Tissue and cell source | Culture approach | Study highlight |
|---|---|---|---|
| Takai et al. [87], 2016 | HCC and hepatoblastoma | 3D culture with porous alginate scaffolds | 3D cultured immortalized HCC cells form glandular epithelial spheroids with increased stemness marker expression compared to cells in monolayer culture. |
| Immortalized HCC cell lines | The 3D cultured HCC cells exhibit greater tumorigenicity and metastasis potential when engrafted in vivo. | ||
| Broutier et al. [35], 2017 | HCC, CCC, cHCC-CCC | ICO culture | The study demonstrated the feasibility of generating tumoroids from all three major liver cancer subtypes using the ICO culture approach. |
| Surgical resections | Tumoroid cultures retain histological architecture and the parental tumor’s mutational and gene expression profile. | ||
| Nuciforo et al. [89], 2018 | HCC and CCC | ICO culture | The study demonstrates that the ICO culture method can also generate tumoroids from pre-surgical needle biopsy samples. |
| Tumor Needle Biopsies | |||
| Saltsman et al. [67], 2020 | Hepatoblastoma | ICO culture | A lack of human models has hindered hepatoblastoma research progress. This study further highlighted the broad utility of the ICO culture approach to generate liver cancer models. |
| Surgical resections | |||
| Li et al. [94], 2019 | HCC and CCC | ICO culture | The study explores the potential of capturing intra-tumor heterogeneity (ITH) across large HCC tumors using the ICO culture method. |
| Multi-sections of a single large resected Tumor | Tumoroids generated from different sectors exhibit significant variation in drug resistance against a panel of 129 drugs. | ||
| Artegiani et al. [96], 2019 | Healthy liver tissue | ICO culture | The study showed that oncogenic transformation of ICO using CRISPR-CAS9 mediated gene-editing enables modeling of cholangiocarcinoma development. |
| Yang et al. [98], 2022 | Healthy fetal liver tissue | Hepatocyte organoid culture | The organoid culture approach enables the stable expansion of hepatocytes isolated from fetal liver tissue. Hippo-YAP activation induces malignant transformation of the fetal hepatocyte organoid into tumoroids resembling fetal hepatoblastoma. |
| Sun et al. [100], 2019 | Human fibroblast | Trans-differentiated hepatic organoids | Overexpression of FOXA3, HNF1A, HNF4A, and SV40 large T antigen induces human fibroblast transdifferentiation to human-induced hepatic organoids (HiHeps). HiHeps can be employed to evaluate oncogenic effects and reveal mechanistic roles of known liver cancer driver genes. |
| Cao et al. [107], 2019 | Tumor from mouse liver cancer models | ICO culture | Tumoroids derived from mouse liver cancer models can be expanded long-term and form tumors when engrafted. Tumors formed recapitulate the heterogeneity of liver cancer observed in cancer patients |
| Cao et al. [95], 2020 | Genetically engineered mouse models (GEMMs) can be used in parallel for investigating the mechanism of cancer initiation and progression. |
Table 3.
| Reference | Genetic disease | Culture approach | Study highlight |
|---|---|---|---|
| Nie et al. [130], 2018 | HBV | Co-culture of PSC-derived hepatic endoderm with MSC and HUVEC | Parallel comparison of PSC-derived hepatocyte-like cells (HLCs) and liver organoids (LOs). LOs are more susceptible to HBV viral infection and could be maintained longer in culture compared to HLC. |
| LO HBV models exhibit cytopathic effects of HBV infection. | |||
| De Crignis et al. [131], 2021 | HBV | ICO culture | HBV-infected organoid models exhibit various disease hallmarks, including cccDNA formation and HBS antigen expression. |
| HBV integration is detectable in the HBV patient-derived organoids and unique gene signatures identified in the transcriptome profiles of HBV patient-derived organoids is enriched in HBV and HCC patient tissue. | |||
| Li et al. [135], 2022 | HEV | ICO culture | Patient-derived fetal and adult organoids can be employed for modeling HEV infection via overexpression of in vitro–transcribed genomic RNA. |
| The organoid model was employed to demonstrate that HEV infects both cholangiocytes and hepatocytes, and viruses were released from the apical surface of both cell types. | |||
| Small-scale antiviral drug screens with the HEV organoids identify Brequinar and Homoharringtonine as potential new HEV therapeutics. | |||
| a. Yang et al. [136], 2020 | SARS-CoV-2 | PSC-derived liver organoids | PSC-derived liver organoid was employed to demonstrate that SARS-CoV-2 virus can infect both hepatocytes and cholangiocyte . |
| b. Richards et al. [137], 2022 | Single-cell transcriptome profiles of infected organoids show that the hepatocytes express higher levels of chemokines and cytokines than the cholangiocytesb. | ||
| a. Yang et al. [136], 2020 | SARS-CoV-2 | Intra-hepatic cholangiocyte organoids | ICO organoid-derived hepatocyte and cholangiocyte organoids can be infected with SARS-CoV-2 virus. Infection induces the expression of immune-related genes in both cell types. |
| b. Zhao et al. [138], 2020; Zhao et al. [139], 2023 | SARS-CoV-2 virus infection results in the downregulation of genes encoding junction proteins and bile transporters in cholangiocyte organoidsa. | ||
| c. McCarron et al. [140], 2021 | ICOs derived from Non-alcoholic steatohepatitis (NASH) patient have increased UBD expression, and exhibits increased sensitivity to SARS-CoV-2 virus infectionC. |
Table 4.
| Reference | Liver disease | Culture approach | Study highlight |
|---|---|---|---|
| Ouchi et al. [58], 2019 | NAFLD | iPSC-derived multi-cellular organoid | PSC-derived hepatic organoids containing both parenchymal and non-parenchymal cells types. |
| When treated with free fatty acids, organoids recapitulate the main features of NAFLD, including steatosis, inflammation, and fibrosis. | |||
| Collin de l’Hortet et al. [180], 2019 | NAFLD | iPSC-derived hepatocytes co-culture with mesenchymal cells, fibroblasts, and macrophages in decellularized rat liver | One of the largest organoids generated, containing multiple liver cell types. Tissue-like organoids enabled NAS scoring similar to patient biopsy samples. |
| A peristaltic pump delivers nutrients and NAFLD-inducing agents to the core of the large organoid to achieve the manifestation of phenotypes throughout the tissue-like culture. | |||
| Kimura et al. [181], 2022 | NAFLD | iPSC-derived multi-cellular organoid | En masse generation of liver organoids from 24 iPSC cell lines in a single matrigel droplet enables high throughput parallel screening for NAFLD phenotype. |
| The high throughput genotype-to-phenotype correlation screen enables rapid validation and potential discovery of functional SNPs. | |||
| Ramli et al. [59], 2020 | NAFLD | iPSC-derived hepatobiliary organoid culture | Hepatobiliary organoids comprise a bile canaliculi network extending from the hepatocyte core to the peripheral cholangiocyte cysts. |
| DILI | The bile canaliculi system can model the cholestasis-inducing side effects of troglitazone. | ||
| The hepatobiliary organoids treated with free fatty acids developed NAFLD phenotypes, including ductular reaction and diminishing bile canaliculi network. | |||
| McCarron et al. [140], 2021 | NAFLD | ICO culture | NASH patient-derived ICO organoids exhibit increased expression of inflammation-associated genes and other genes associated with fibrogenesis and tumorigenesis. |
| Idiopathic responses were observed across organoids generated from different patients. | |||
| Wang et al. [182], 2019 | ALD | PSC-derived bipotent epithelial organoid | PSC-derived expandable hepatic organoids (EHO) can differentiate into functional hepatocytes or bile duct cells. |
| Co-culture and differentiation of EHO with mesenchymal cells generate functional liver organoids that can model ALD pathophysiology after ethanol treatment, including oxidative stress, steatosis, inflammation, and fibrosis. | |||
| Shinozawa et al. [57], 2021 | DILI | iPSC-derived hepatocyte organoid | Organoids comprise mainly hepatocytes with bile canaliculi-like architecture that supports unidirectional bile acid transport. |
| The authors developed an oOrganoid-based drug toxicity assay with multiple readout measurements for cell viability, cholestasis, and mitochondrial toxicity. The organoid model showed high predictive value for 238 clinically available drugs. | |||
| The study further demonstrated the organoid models’ utility for evaluating the pharmacogenomics effect of Bosentan-induced Cholestasis. |
- Abbreviations
- Abbreviations
NPCs nonparenchymal liver cells
HSCs hepatic stellate cells
KCs Kupffer cells
DILI drug-induced liver injury
CLD chronic liver disease
PLC primary liver cancer
NAFLD nonalcoholic fatty liver disease
ALD alcoholic liver disease
ECM extracellular matrix
PSC pluripotent stem cell
iPSC induced pluripotent stem cell
PHHs primary human hepatocytes
HCC hepatocellular carcinoma
ICOs intrahepatic cholangiocyte organoids
A1AT a1-antitrypsin
ALGS Alagille syndrome
GEMMs genetically engineered mouse models
CLC cholangiocyte-like cell
CF cystic fibrosis
CFTR cystic fibrosis transmembrane conductance regulator
HLCs hepatocyte-like cells
AFP alpha-fetal protein
CCC cholangiocarcinoma
cHCC-CCC combined hepatocellular cholangiocarcinoma
HBV hepatitis B virus
HCV hepatitis C virus
NASH nonalcoholic steatohepatitis
HOs hepatocyte organoids
NRTIs nucleoside reverse transcriptase inhibitors
NTCP sodium taurocholate co-transporting peptide
DAA direct-acting antiviral
LOs liver organoids
HOs healthy organoids
DOs disease organoids
SARS-CoV-2 severe acute respiratory syndrome coronavirus 2
SNPs single-nucleotide polymorphisms
GCKR glucokinase regulator
PNPLA3 patatin-like phospholipase domain containing 3
FFAs free fatty acids
TNF-α tumor necrosis factor-alpha
IL-6 interleukin-6
DR ductular reaction
- REFERENCES
- REFERENCES
REFERENCES
1. Trefts E, Gannon M, Wasserman DH. The liver. Curr Biol 2017;27:R1147-R1151.
[Article] [PubMed] [PMC]2. Schulze RJ, Schott MB, Casey CA, Tuma PL, McNiven MA. The cell biology of the hepatocyte: A membrane trafficking machine. J Cell Biol 2019;218:2096-2112.
[Article] [PubMed] [PMC]3. Zhou Z, Xu MJ, Gao B. Hepatocytes: a key cell type for innate immunity. Cell Mol Immunol 2016;13:301-315.
[Article] [PubMed] [PMC]4. Malik R, Selden C, Hodgson H. The role of non-parenchymal cells in liver growth. Semin Cell Dev Biol 2002;13:425-431.
[Article] [PubMed]5. Kumar S, Duan Q, Wu R, Harris EN, Su Q. Pathophysiological communication between hepatocytes and non-parenchymal cells in liver injury from NAFLD to liver fibrosis. Adv Drug Deliv Rev 2021;176:113869.
[Article] [PubMed]6. Cheemerla S, Balakrishnan M. Global epidemiology of chronic liver disease. Clin Liver Dis (Hoboken) 2021;17:365-370.
[Article] [PubMed] [PMC]7. Moon AM, Singal AG, Tapper EB. Contemporary epidemiology of chronic liver disease and cirrhosis. Clin Gastroenterol Hepatol 2020;18:2650-2666.
[Article] [PubMed] [PMC]8. Yu JH, Ma S. Organoids as research models for hepatocellular carcinoma. Exp Cell Res 2022;411:112987.
[Article] [PubMed]9. Liu Y, Meyer C, Xu C, Weng H, Hellerbrand C, ten Dijke P, et al. Animal models of chronic liver diseases. Am J Physiol Gastrointest Liver Physiol 2013;304:G449-G468.
[Article] [PubMed]10. Ortiz C, Schierwagen R, Schaefer L, Klein S, Trepat X, Trebicka J. Extracellular matrix remodeling in chronic liver disease. Curr Tissue Microenviron Rep 2021;2:41-52.
[Article] [PubMed] [PMC]11. Simian M, Bissell MJ. Organoids: A historical perspective of thinking in three dimensions. J Cell Biol 2017;216:31-40.
[Article] [PubMed] [PMC]12. Corrò C, Novellasdemunt L, Li VSW. A brief history of organoids. Am J Physiol Cell Physiol 2020;319:C151-C165.
[Article] [PubMed] [PMC]13. Evans MJ, Kaufman MH. Establishment in culture of pluripotential cells from mouse embryos. Nature 1981;292:154-156.
[Article] [PubMed]14. Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, et al. Embryonic stem cell lines derived from human blastocysts. Science 1998;282:1145-1147. Erratum in: Science 1998;282:1827.15. Kim J, Koo BK, Knoblich JA. Human organoids: model systems for human biology and medicine. Nat Rev Mol Cell Biol 2020;21:571-584.
[Article] [PubMed] [PMC]16. Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, et al. Cerebral organoids model human brain development and microcephaly. Nature 2013;501:373-379.
[Article] [PubMed] [PMC]17. Dye BR, Hill DR, Ferguson MA, Tsai YH, Nagy MS, Dyal R, et al. In vitro generation of human pluripotent stem cell derived lung organoids. Elife 2015;4:e05098.
[Article] [PubMed] [PMC]18. Takasato M, Er PX, Chiu HS, Maier B, Baillie GJ, Ferguson C, et al. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature 2015;526:564-568. Erratum in: Nature 2016;536:238.19. McCracken KW, Catá EM, Crawford CM, Sinagoga KL, Schumacher M, Rockich BE, et al. Modelling human development and disease in pluripotent stem-cell-derived gastric organoids. Nature 2014;516:400-404.
[Article] [PubMed] [PMC]20. Spence JR, Mayhew CN, Rankin SA, Kuhar MF, Vallance JE, Tolle K, et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 2011;470:105-109.
[Article] [PubMed] [PMC]21. Breunig M, Merkle J, Wagner M, Melzer MK, Barth TFE, Engleitner T, et al. Modeling plasticity and dysplasia of pancreatic ductal organoids derived from human pluripotent stem cells. Cell Stem Cell 2021;28:1105-1124.e19.
[Article] [PubMed] [PMC]22. Mun SJ, Ryu JS, Lee MO, Son YS, Oh SJ, Cho HS, et al. Generation of expandable human pluripotent stem cell-derived hepatocyte-like liver organoids. J Hepatol 2019;71:970-985.
[Article] [PubMed]23. Takebe T, Sekine K, Enomura M, Koike H, Kimura M, Ogaeri T, et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 2013;499:481-484.
[Article] [PubMed]24. Rowe RG, Daley GQ. Induced pluripotent stem cells in disease modelling and drug discovery. Nat Rev Genet 2019;20:377-388.
[Article] [PubMed] [PMC]25. Li ML, Aggeler J, Farson DA, Hatier C, Hassell J, Bissell MJ. Influence of a reconstituted basement membrane and its components on casein gene expression and secretion in mouse mammary epithelial cells. Proc Natl Acad Sci U S A 1987;84:136-140.
[Article] [PubMed] [PMC]26. Shannon JM, Mason RJ, Jennings SD. Functional differentiation of alveolar type II epithelial cells in vitro: effects of cell shape, cell-matrix interactions and cell-cell interactions. Biochim Biophys Acta 1987;931:143-156.
[Article] [PubMed]27. Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009;459:262-265.
[Article] [PubMed]28. Hu H, Gehart H, Artegiani B, LÖpez-Iglesias C, Dekkers F, Basak O, et al. Long-term expansion of functional mouse and human hepatocytes as 3D organoids. Cell 2018;175:1591-1606.e19.
[Article] [PubMed]29. Sato T, Stange DE, Ferrante M, Vries RG, Van Es JH, Van den Brink S, et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011;141:1762-1772.
[Article] [PubMed]30. Bartfeld S, Bayram T, van de Wetering M, Huch M, Begthel H, Kujala P, et al. In vitro expansion of human gastric epithelial stem cells and their responses to bacterial infection. Gastroenterology 2015;148:126-136.e6.
[Article] [PubMed] [PMC]31. Yee C, Dickson KA, Muntasir MN, Ma Y, Marsh DJ. Three-dimensional modelling of ovarian cancer: From cell lines to organoids for discovery and personalized medicine. Front Bioeng Biotechnol 2022;10:836984.
[Article] [PubMed] [PMC]32. van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A, et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015;161:933-945.
[Article] [PubMed] [PMC]33. Boj SF, Hwang CI, Baker LA, Chio II, Engle DD, Corbo V, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015;160:324-338.
[Article] [PubMed] [PMC]34. Li X, Francies HE, Secrier M, Perner J, Miremadi A, Galeano-Dalmau N, et al. Organoid cultures recapitulate esophageal adenocarcinoma heterogeneity providing a model for clonality studies and precision therapeutics. Nat Commun 2018;9:2983.
[Article] [PubMed] [PMC]35. Broutier L, Mastrogiovanni G, Verstegen MM, Francies HE, Gavarró LM, Bradshaw CR, et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat Med 2017;23:1424-1435.
[Article] [PubMed] [PMC]36. Martínez AK, Maroni L, Marzioni M, Ahmed ST, Milad M, Ray D, et al. Mouse models of liver fibrosis mimic human liver fibrosis of different etiologies. Curr Pathobiol Rep 2014;2:143-153.
[Article] [PubMed] [PMC]37. Fiorotto R, Amenduni M, Mariotti V, Fabris L, Spirli C, Strazzabosco M. Liver diseases in the dish: iPSC and organoids as a new approach to modeling liver diseases. Biochim Biophys Acta Mol Basis Dis 2019;1865:920-928.
[Article] [PubMed] [PMC]38. Vinken M. Primary hepatocyte cultures for liver disease modeling. Curr Opin Toxicol 2021;25:1-5.
[Article]39. Arzumanian VA, Kiseleva OI, Poverennaya EV. The curious case of the HepG2 cell line: 40 years of expertise. Int J Mol Sci 2021;22:13135.
[Article] [PubMed] [PMC]40. Nuciforo S, Heim MH. Organoids to model liver disease. JHEP Rep 2020;3:100198.
[Article] [PubMed] [PMC]41. Lam DTUH, Dan YY, Chan YS, Ng HH. Emerging liver organoid platforms and technologies. Cell Regen 2021;10:27.
[Article] [PubMed] [PMC]42. Marsee A, Roos FJM, Verstegen MMA; HPB Organoid Consortium, Gehart H, de Koning E, et al. Building consensus on definition and nomenclature of hepatic, pancreatic, and biliary organoids. Cell Stem Cell 2021;28:816-832.
[Article] [PubMed]43. Fagiuoli S, Daina E, D’Antiga L, Colledan M, Remuzzi G. Monogenic diseases that can be cured by liver transplantation. J Hepatol 2013;59:595-612.
[Article] [PubMed]44. Lachmann RH. Enzyme replacement therapy for lysosomal storage diseases. Curr Opin Pediatr 2011;23:588-593.
[Article] [PubMed]45. Walshe JM, Yealland M. Chelation treatment of neurological Wilson’s disease. Q J Med 1993;86:197-204.
[PubMed]46. Boudes PF. Gene therapy as a new treatment option for inherited monogenic diseases. Eur J Intern Med 2014;25:31-36.
[Article] [PubMed]47. Huch M, Gehart H, van Boxtel R, Hamer K, Blokzijl F, Verstegen MM, et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell 2015;160:299-312.
[Article] [PubMed] [PMC]48. Caiazza C, Parisi S, Caiazzo M. Liver organoids: Updates on disease modeling and biomedical applications. Biology (Basel) 2021;10:835.
[Article] [PubMed] [PMC]49. Gómez-Mariano G, Matamala N, Martínez S, Justo I, Marcacuzco A, Jimenez C, et al. Liver organoids reproduce alpha-1 antitrypsin deficiency-related liver disease. Hepatol Int 2020;14:127-137.
[Article] [PubMed] [PMC]50. Fromme M, Schneider CV, Trautwein C, Brunetti-Pierri N, Strnad P. Alpha-1 antitrypsin deficiency: A re-surfacing adult liver disorder. J Hepatol 2022;76:946-958.
[Article] [PubMed]51. Andersson ER, Chivukula IV, Hankeova S, Sjöqvist M, Tsoi YL, Ramsköld D, et al. Mouse model of alagille syndrome and mechanisms of Jagged1 missense mutations. Gastroenterology 2018;154:1080-1095.
[Article] [PubMed] [PMC]52. Kruitwagen HS, Oosterhoff LA, van Wolferen ME, Chen C, Nantasanti Assawarachan S, Schneeberger K, et al. Long-term survival of transplanted autologous canine liver organoids in a COMMD1-deficient dog model of metabolic liver disease. Cells 2020;9:410.
[Article] [PubMed] [PMC]53. Nantasanti S, Spee B, Kruitwagen HS, Chen C, Geijsen N, Oosterhoff LA, et al. Disease modeling and gene therapy of copper storage disease in canine hepatic organoids. Stem Cell Reports 2015;5:895-907.
[Article] [PubMed] [PMC]54. Shi Y, Inoue H, Wu JC, Yamanaka S. Induced pluripotent stem cell technology: a decade of progress. Nat Rev Drug Discov 2017;16:115-130.
[Article] [PubMed] [PMC]55. Ogawa M, Ogawa S, Bear CE, Ahmadi S, Chin S, Li B, et al. Directed differentiation of cholangiocytes from human pluripotent stem cells. Nat Biotechnol 2015;33:853-861.
[Article] [PubMed]56. Sampaziotis F, de Brito MC, Madrigal P, Bertero A, Saeb-Parsy K, Soares FAC, et al. Cholangiocytes derived from human induced pluripotent stem cells for disease modeling and drug validation. Nat Biotechnol 2015;33:845-852.
[Article] [PubMed] [PMC]57. Shinozawa T, Kimura M, Cai Y, Saiki N, Yoneyama Y, Ouchi R, et al. High-fidelity drug-induced liver injury screen using human pluripotent stem cell-derived organoids. Gastroenterology 2021;160:831-846.e10.
[Article] [PubMed] [PMC]58. Ouchi R, Togo S, Kimura M, Shinozawa T, Koido M, Koike H, et al. Modeling steatohepatitis in humans with pluripotent stem cell-derived organoids. Cell Metab 2019;30:374-384.e6.
[Article] [PubMed] [PMC]59. Ramli MNB, Lim YS, Koe CT, Demircioglu D, Tng W, Gonzales KAU, et al. Human pluripotent stem cell-derived organoids as models of liver disease. Gastroenterology 2020;159:1471-1486.e12.
[Article] [PubMed]60. Shteinberg M, Haq IJ, Polineni D, Davies JC. Cystic fibrosis. Lancet 2021;397:2195-2211.
[Article] [PubMed]61. Guan Y, Xu D, Garfin PM, Ehmer U, Hurwitz M, Enns G, et al. Human hepatic organoids for the analysis of human genetic diseases. JCI Insight 2017;2:e94954.
[Article] [PubMed] [PMC]62. Akbari S, Sevinç GG, Ersoy N, Basak O, Kaplan K, Sevinç K, et al. Robust, long-term culture of endoderm-derived hepatic organoids for disease modeling. Stem Cell Reports 2019;13:627-641.
[Article] [PubMed] [PMC]63. Graffmann N, Scherer B, Adjaye J. In vitro differentiation of pluripotent stem cells into hepatocyte like cells - Basic principles and current progress. Stem Cell Res 2022;61:102763.
[Article] [PubMed]64. Kvist AJ, Kanebratt KP, Walentinsson A, Palmgren H, O’Hara M, Björkbom A, et al. Critical differences in drug metabolic properties of human hepatic cellular models, including primary human hepatocytes, stem cell derived hepatocytes, and hepatoma cell lines. Biochem Pharmacol 2018;155:124-140.
[Article] [PubMed]65. Ma H, de Zwaan E, Guo YE, Cejas P, Thiru P, van de Bunt M, et al. The nuclear receptor THRB facilitates differentiation of human PSCs into more mature hepatocytes. Cell Stem Cell 2022;29:795-809.e11. Erratum in: Cell Stem Cell 2022;29:1611.66. Yang J, Pan G, Guan L, Liu Z, Wu Y, Liu Z, et al. The burden of primary liver cancer caused by specific etiologies from 1990 to 2019 at the global, regional, and national levels. Cancer Med 2022;11:1357-1370.
[Article] [PubMed] [PMC]67. Saltsman JA, Hammond WJ, Narayan NJC, Requena D, Gehart H, Lalazar G, et al. A human organoid model of aggressive hepatoblastoma for disease modeling and drug testing. Cancers (Basel) 2020;12:2668.
[Article] [PubMed] [PMC]68. Lin J, Zhang H, Yu H, Bi X, Zhang W, Yin J, et al. Epidemiological characteristics of primary liver cancer in mainland China from 2003 to 2020: A representative multicenter study. Front Oncol 2022;12:906778.
[Article] [PubMed] [PMC]69. Piñero F, Pages J, Marciano S, Fernández N, Silva J, Anders M, et al. Fatty liver disease, an emerging etiology of hepatocellular carcinoma in Argentina. World J Hepatol 2018;10:41-50.
[Article] [PubMed] [PMC]70. Teschke R. Hepatocellular carcinoma in alcoholic liver disease: mechanistic considerations and clinical facts. Hepatoma Res 2019;5:40.
[Article]71. Al-Salama ZT, Syed YY, Scott LJ. Lenvatinib: A review in hepatocellular carcinoma. Drugs 2019;79:665-674.
[Article] [PubMed]72. Personeni N, Pressiani T, Rimassa L. Lenvatinib for the treatment of unresectable hepatocellular carcinoma: evidence to date. J Hepatocell Carcinoma 2019;6:31-39.
[PubMed] [PMC]73. Madden K, Kasler MK. Immune checkpoint inhibitors in lung cancer and melanoma. Semin Oncol Nurs 2019;35:150932.
[Article] [PubMed]74. Feng GS, Hanley KL, Liang Y, Lin X. Improving the efficacy of liver cancer immunotherapy: The power of combined preclinical and clinical studies. Hepatology 2021;73 Suppl 1:104-114.
[Article] [PubMed] [PMC]75. Marquardt JU, Andersen JB, Thorgeirsson SS. Functional and genetic deconstruction of the cellular origin in liver cancer. Nat Rev Cancer 2015;15:653-667.
[Article] [PubMed]76. Ding X, He M, Chan AWH, Song QX, Sze SC, et al. Genomic and epigenomic features of primary and recurrent hepatocellular carcinomas. Gastroenterology 2019;157:1630-1645.e6.
[Article] [PubMed]77. Harding JJ, Nandakumar S, Armenia J, Khalil DN, Albano M, Ly M, et al. Prospective genotyping of hepatocellular carcinoma: Clinical implications of next-generation sequencing for matching patients to targeted and immune therapies. Clin Cancer Res 2019;25:2116-2126.
[Article] [PubMed] [PMC]78. Lin DC, Mayakonda A, Dinh HQ, Huang P, Lin L, Liu X, et al. Genomic and epigenomic heterogeneity of hepatocellular carcinoma. Cancer Res 2017;77:2255-2265.
[Article] [PubMed] [PMC]79. Okrah K, Tarighat S, Liu B, Koeppen H, Wagle MC, Cheng G, et al. Transcriptomic analysis of hepatocellular carcinoma reveals molecular features of disease progression and tumor immune biology. NPJ Precis Oncol 2018;2:25.
[Article] [PubMed] [PMC]80. Rebouissou S, Franconi A, Calderaro J, Letouzé E, Imbeaud S, Pilati C, et al. Genotype-phenotype correlation of CTNNB1 mutations reveals different ß-catenin activity associated with liver tumor progression. Hepatology 2016;64:2047-2061.
[Article] [PubMed]81. Ishii T, Tamura A, Shibata T, Kuroda K, Kanda T, Sugiyama M, et al. Analysis of HBV genomes integrated into the genomes of human hepatoma PLC/PRF/5 cells by HBV sequence capture-based next-generation sequencing. Genes (Basel) 2020;11:661.
[Article] [PubMed] [PMC]82. Kamatani Y, Wattanapokayakit S, Ochi H, Kawaguchi T, Takahashi A, Hosono N, et al. A genome-wide association study identifies variants in the HLA-DP locus associated with chronic hepatitis B in Asians. Nat Genet 2009;41:591-595.
[Article] [PubMed]83. Sze KM, Ho DW, Chiu YT, Tsui YM, Chan LK, Lee JM, et al. Hepatitis B Virus-telomerase reverse transcriptase promoter integration harnesses host ELF4, resulting in telomerase reverse transcriptase gene transcription in hepatocellular carcinoma. Hepatology 2021;73:23-40.
[Article] [PubMed] [PMC]84. Byrne CD, Targher G. NAFLD: a multisystem disease. J Hepatol 2015;62(1 Suppl):S47-S64.
[Article] [PubMed]85. Curtius K, Wright NA, Graham TA. An evolutionary perspective on field cancerization. Nat Rev Cancer 2018;18:19-32.
[Article] [PubMed]86. Greten FR, Grivennikov SI. Inflammation and cancer: Triggers, mechanisms, and consequences. Immunity 2019;51:27-41.
[Article] [PubMed] [PMC]87. Takai A, Fako V, Dang H, Forgues M, Yu Z, Budhu A, et al. Three-dimensional organotypic culture models of human hepatocellular carcinoma. Sci Rep 2016;6:21174.
[Article] [PubMed] [PMC]88. He X, Hikiba Y, Suzuki Y, Nakamori Y, Kanemaru Y, Sugimori M, et al. EGFR inhibition reverses resistance to lenvatinib in hepatocellular carcinoma cells. Sci Rep 2022;12:8007.
[Article] [PubMed] [PMC]89. Nuciforo S, Fofana I, Matter MS, Blumer T, Calabrese D, Boldanova T, et al. Organoid models of human liver cancers derived from tumor needle biopsies. Cell Rep 2018;24:1363-1376.
[Article] [PubMed] [PMC]90. Xu H, Lyu X, Yi M, Zhao W, Song Y, Wu K. Organoid technology and applications in cancer research. J Hematol Oncol 2018;11:116.
[Article] [PubMed] [PMC]91. Wu LJ, Chen ZY, Wang Y, Zhao JG, Xie XZ, Chen G. Organoids of liver diseases: From bench to bedside. World J Gastroenterol 2019;25:1913-1927.
[Article] [PubMed] [PMC]92. El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology 2007;132:2557-2576.
[Article] [PubMed]93. Zhou C, Zhang W, Chen W, Yin Y, Atyah M, Liu S, et al. Integrated analysis of copy number variations and gene expression profiling in hepatocellular carcinoma. Sci Rep 2017;7:10570.
[Article] [PubMed] [PMC]94. Li L, Knutsdottir H, Hui K, Weiss MJ, He J, Philosophe B, et al. Human primary liver cancer organoids reveal intratumor and interpatient drug response heterogeneity. JCI Insight 2019;4:e121490.
[Article] [PubMed] [PMC]95. Cao W, Li M, Liu J, Zhang S, Noordam L, Verstegen MMA, et al. LGR5 marks targetable tumor-initiating cells in mouse liver cancer. Nat Commun 2020;11:1961.
[Article] [PubMed] [PMC]96. Artegiani B, van Voorthuijsen L, Lindeboom RGH, Seinstra D, Heo I, Tapia P, et al. Probing the tumor suppressor function of BAP1 in CRISPR-engineered human liver organoids. Cell Stem Cell 2019;24:927-943.e6.
[Article] [PubMed]97. Chan-On W, Nairismägi ML, Ong CK, Lim WK, Dima S, Pairojkul C, et al. Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nat Genet 2013;45:1474-1478.
[Article] [PubMed]98. Yang L, Chen J, Liang J, Zhang Y, Wang Q, Ren X, et al. Modeling hepatoblastoma development with human fetal liver organoids reveals YAP1 activation is sufficient for tumorigenesis. Protein Cell 2022;13:683-688.
[Article] [PubMed] [PMC]99. Huch M, Dorrell C, Boj SF, van Es JH, Li VS, van de Wetering M, et al. In vitro expansion of single Lgr5+ liver stem cells induced by Wnt-driven regeneration. Nature 2013;494:247-250.
[Article] [PubMed] [PMC]100. Sun L, Wang Y, Cen J, Ma X, Cui L, Qiu Z, et al. Modelling liver cancer initiation with organoids derived from directly reprogrammed human hepatocytes. Nat Cell Biol 2019;21:1015-1026.
[Article] [PubMed]101. Huang P, Zhang L, Gao Y, He Z, Yao D, Wu Z, et al. Direct reprogramming of human fibroblasts to functional and expandable hepatocytes. Cell Stem Cell 2014;14:370-384.
[Article] [PubMed]102. Zheng K, Cubero FJ, Nevzorova YA. c-MYC-making liver sick: Role of c-MYC in hepatic cell function, homeostasis and disease. Genes (Basel) 2017;8:123.
[Article] [PubMed] [PMC]103. Sun L, Hui L. Progress in human liver organoids. J Mol Cell Biol 2020;12:607-617.
[Article] [PubMed] [PMC]104. Dijkstra KK, Monkhorst K, Schipper LJ, Hartemink KJ, Smit EF, Kaing S, et al. Challenges in establishing pure lung cancer organoids limit their utility for personalized medicine. Cell Rep 2020;31:107588.
[Article] [PubMed]105. Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, Gopalan A, et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 2014;159:176-187.
[Article] [PubMed] [PMC]106. Fujii M, Shimokawa M, Date S, Takano A, Matano M, Nanki K, et al. A colorectal tumor organoid library demonstrates progressive loss of niche factor requirements during tumorigenesis. Cell Stem Cell 2016;18:827-838.
[Article] [PubMed]107. Cao W, Liu J, Wang L, Li M, Verstegen MMA, Yin Y, et al. Modeling liver cancer and therapy responsiveness using organoids derived from primary mouse liver tumors. Carcinogenesis 2019;40:145-154.
[Article] [PubMed]108. Protzer U, Maini MK, Knolle PA. Living in the liver: hepatic infections. Nat Rev Immunol 2012;12:201-213.
[Article] [PubMed]109. World Health Organization. Global hepatitis report 2017. Geneva: World Health Organization; 2017.110. Fanning GC, Zoulim F, Hou J, Bertoletti A. Therapeutic strategies for hepatitis B virus infection: towards a cure. Nat Rev Drug Discov 2019;18:827-844. Erratum in: Nat Rev Drug Discov 2020;19:291.111. Roca Suarez AA, Testoni B, Zoulim F. HBV 2021: New therapeutic strategies against an old foe. Liver Int 2021;41 Suppl 1:15-23.
[PubMed]112. Degasperi E, Anolli MP, Lampertico P. Bulevirtide for patients with compensated chronic hepatitis delta: A review. Liver Int 2022 Aug 9;doi: 10.1111/liv.15389.
[Article] [PubMed]113. Nassal M. HBV cccDNA: viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 2015;64:1972-1984.
[Article] [PubMed]114. Manns MP, Maasoumy B. Breakthroughs in hepatitis C research: from discovery to cure. Nat Rev Gastroenterol Hepatol 2022;19:533-550.
[Article] [PubMed] [PMC]115. Kolykhalov AA, Agapov EV, Blight KJ, Mihalik K, Feinstone SM, Rice CM. Transmission of hepatitis C by intrahepatic inoculation with transcribed RNA. Science 1997;277:570-574.
[Article] [PubMed]116. Yanagi M, Purcell RH, Emerson SU, Bukh J. Transcripts from a single full-length cDNA clone of hepatitis C virus are infectious when directly transfected into the liver of a chimpanzee. Proc Natl Acad Sci U S A 1997;94:8738-8743.
[Article] [PubMed] [PMC]117. Baktash Y, Madhav A, Coller KE, Randall G. Single particle imaging of polarized hepatoma organoids upon hepatitis C virus infection reveals an ordered and sequential entry process. Cell Host Microbe 2018;23:382-394.e5.
[Article] [PubMed] [PMC]118. Ko C, Lee S, Windisch MP, Ryu WS. DDX3 DEAD-box RNA helicase is a host factor that restricts hepatitis B virus replication at the transcriptional level. J Virol 2014;88:13689-13698.
[Article] [PubMed] [PMC]119. Nishitsuji H, Harada K, Ujino S, Zhang J, Kohara M, Sugiyama M, et al. Investigating the hepatitis B virus life cycle using engineered reporter hepatitis B viruses. Cancer Sci 2018;109:241-249.
[Article] [PubMed] [PMC]120. Verrier ER, Colpitts CC, Bach C, Heydmann L, Weiss A, Renaud M, et al. A targeted functional RNA interference screen uncovers glypican 5 as an entry factor for hepatitis B and D viruses. Hepatology 2016;63:35-48.
[Article] [PubMed]121. Lucifora J, Durantel D, Testoni B, Hantz O, Levrero M, Zoulim F. Control of hepatitis B virus replication by innate response of HepaRG cells. Hepatology 2010;51:63-72.
[Article] [PubMed]122. Macovei A, Radulescu C, Lazar C, Petrescu S, Durantel D, Dwek RA, et al. Hepatitis B virus requires intact caveolin-1 function for productive infection in HepaRG cells. J Virol 2010;84:243-253.
[Article] [PubMed] [PMC]123. Ni Y, Lempp FA, Mehrle S, Nkongolo S, Kaufman C, Fälth M, et al. Hepatitis B and D viruses exploit sodium taurocholate cotransporting polypeptide for species-specific entry into hepatocytes. Gastroenterology 2014;146:1070-1083.
[Article] [PubMed]124. Schulze A, Gripon P, Urban S. Hepatitis B virus infection initiates with a large surface protein-dependent binding to heparan sulfate proteoglycans. Hepatology 2007;46:1759-1768.
[Article] [PubMed]125. Sureau C, Salisse J. A conformational heparan sulfate binding site essential to infectivity overlaps with the conserved hepatitis B virus a-determinant. Hepatology 2013;57:985-994.
[Article] [PubMed]126. Hantz O, Parent R, Durantel D, Gripon P, Guguen-Guillouzo C, Zoulim F. Persistence of the hepatitis B virus covalently closed circular DNA in HepaRG human hepatocyte-like cells. J Gen Virol 2009;90:127-135.
[Article] [PubMed]127. Sakurai F, Mitani S, Yamamoto T, Takayama K, Tachibana M, Watashi K, et al. Human induced-pluripotent stem cell-derived hepatocyte-like cells as an in vitro model of human hepatitis B virus infection. Sci Rep 2017;7:45698.
[Article] [PubMed] [PMC]128. Shlomai A, Schwartz RE, Ramanan V, Bhatta A, de Jong YP, Bhatia SN, et al. Modeling host interactions with hepatitis B virus using primary and induced pluripotent stem cell-derived hepatocellular systems. Proc Natl Acad Sci U S A 2014;111:12193-12198.
[Article] [PubMed] [PMC]129. Xia Y, Carpentier A, Cheng X, Block PD, Zhao Y, Zhang Z, et al. Human stem cell-derived hepatocytes as a model for hepatitis B virus infection, spreading and virus-host interactions. J Hepatol 2017;66:494-503.
[Article] [PubMed] [PMC]130. Nie YZ, Zheng YW, Miyakawa K, Murata S, Zhang RR, Sekine K, et al. Recapitulation of hepatitis B virus-host interactions in liver organoids from human induced pluripotent stem cells. EBioMedicine 2018;35:114-123.
[Article] [PubMed] [PMC]131. De Crignis E, Hossain T, Romal S, Carofiglio F, Moulos P, Khalid MM, et al. Application of human liver organoids as a patient-derived primary model for HBV infection and related hepatocellular carcinoma. Elife 2021;10:e60747.
[PubMed] [PMC]132. Medhat A, Arzumanyan A, Feitelson MA. Hepatitis B x antigen (HBx) is an important therapeutic target in the pathogenesis of hepatocellular carcinoma. Oncotarget 2021;12:2421-2433.
[Article] [PubMed] [PMC]133. Ma XX, Ji Y, Jin L, Baloch Z, Zhang DR, Wang Y, et al. Prevalence and clinical features of hepatitis E virus infection in pregnant women: A large cohort study in Inner Mongolia, China. Clin Res Hepatol Gastroenterol 2021;45:101536.
[Article] [PubMed]134. Kinast V, Burkard TL, Todt D, Steinmann E. Hepatitis E virus drug development. Viruses 2019;11:485.
[Article] [PubMed] [PMC]135. Li P, Li Y, Wang Y, Liu J, Lavrijsen M, Li Y, et al. Recapitulating hepatitis E virus-host interactions and facilitating antiviral drug discovery in human liver-derived organoids. Sci Adv 2022;8:eabj5908.
[Article] [PubMed] [PMC]136. Yang L, Han Y, Nilsson-Payant BE, Gupta V, Wang P, Duan X, et al. A human pluripotent stem cell-based platform to study SARS-CoV-2 tropism and model virus infection in human cells and organoids. Cell Stem Cell 2020;27:125-136.e7.
[Article] [PubMed] [PMC]137. Richards A, Friesen M, Khalil A, Barrasa MI, Gehrke L, Jaenisch R. SARS-CoV-2 infection of human pluripotent stem cell-derived liver organoids reveals potential mechanisms of liver pathology. iScience 2022;25:105146.
[Article] [PubMed] [PMC]138. Zhao B, Ni C, Gao R, Wang Y, Yang L, Wei J, et al. Recapitulation of SARS-CoV-2 infection and cholangiocyte damage with human liver ductal organoids. Protein Cell 2020;11:771-775.
[Article] [PubMed] [PMC]139. Zhao Y, Ren X, Lu J, He M, Liu Z, Yi L, et al. Mechanistic insight of SARS-CoV-2 infection using human hepatobiliary organoids. Gut 2023;72:216-218.
[Article] [PubMed] [PMC]140. McCarron S, Bathon B, Conlon DM, Abbey D, Rader DJ, Gawronski K, et al. Functional characterization of organoids derived from irreversibly damaged liver of patients with NASH. Hepatology 2021;74:1825-1844.
[Article] [PubMed]141. Lagana SM, Kudose S, Iuga AC, Lee MJ, Fazlollahi L, Remotti HE, et al. Hepatic pathology in patients dying of COVID-19: a series of 40 cases including clinical, histologic, and virologic data. Mod Pathol 2020;33:2147-2155.
[Article] [PubMed] [PMC]142. Wang Y, Liu S, Liu H, Li W, Lin F, Jiang L, et al. SARS-CoV-2 infection of the liver directly contributes to hepatic impairment in patients with COVID-19. J Hepatol 2020;73:807-816.
[Article] [PubMed] [PMC]143. Weber S, Hellmuth JC, Scherer C, Muenchhoff M, Mayerle J, Gerbes AL. Liver function test abnormalities at hospital admission are associated with severe course of SARS-CoV-2 infection: a prospective cohort study. Gut 2021;70:1925-1932.
[Article] [PubMed] [PMC]144. Muus C, Luecken MD, Eraslan G, Sikkema L, Waghray A, Heimberg G, et al.; NHLBI LungMap Consortium; Human Cell Atlas Lung Biological Network. Single-cell meta-analysis of SARS-CoV-2 entry genes across tissues and demographics. Nat Med 2021;27:546-559.
[PubMed] [PMC]145. Ji D, Qin E, Xu J, Zhang D, Cheng G, Wang Y, et al. Non-alcoholic fatty liver diseases in patients with COVID-19: A retrospective study. J Hepatol 2020;73:451-453.
[Article] [PubMed] [PMC]146. Loomba R, Sanyal AJ. The global NAFLD epidemic. Nat Rev Gastroenterol Hepatol 2013;10:686-690.
[Article] [PubMed]147. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016;64:73-84.
[Article] [PubMed]148. Boeckmans J, Natale A, Buyl K, Rogiers V, De Kock J, Vanhaecke T, et al. Human-based systems: Mechanistic NASH modelling just around the corner? Pharmacol Res 2018;134:257-267.
[Article] [PubMed]149. Li Z, Xue J, Chen P, Chen L, Yan S, Liu L. Prevalence of nonalcoholic fatty liver disease in mainland of China: a meta-analysis of published studies. J Gastroenterol Hepatol 2014;29:42-51.
[Article] [PubMed]150. Estes C, Anstee QM, Arias-Loste MT, Bantel H, Bellentani S, Caballeria J, et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016-2030. J Hepatol 2018;69:896-904.
[Article] [PubMed]151. Wong RJ, Aguilar M, Cheung R, Perumpail RB, Harrison SA, Younossi ZM, et al. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 2015;148:547-555.
[Article] [PubMed]152. Berná G, Romero-Gomez M. The role of nutrition in non-alcoholic fatty liver disease: Pathophysiology and management. Liver Int 2020;40 Suppl 1:102-108.
[PubMed]153. Bazick J, Donithan M, Neuschwander-Tetri BA, Kleiner D, Brunt EM, Wilson L, et al. Clinical model for NASH and advanced fibrosis in adult patients with diabetes and NAFLD: Guidelines for referral in NAFLD. Diabetes Care 2015;38:1347-1355.
[Article] [PubMed] [PMC]154. Huang PL. A comprehensive definition for metabolic syndrome. Dis Model Mech 2009;2:231-237.
[Article] [PubMed] [PMC]155. Portillo-Sanchez P, Bril F, Maximos M, Lomonaco R, Biernacki D, Orsak B, et al. High prevalence of nonalcoholic fatty liver disease in patients with type 2 diabetes mellitus and normal plasma aminotransferase levels. J Clin Endocrinol Metab 2015;100:2231-2238.
[Article] [PubMed] [PMC]156. Schwimmer JB, Celedon MA, Lavine JE, Salem R, Campbell N, Schork NJ, et al. Heritability of nonalcoholic fatty liver disease. Gastroenterology 2009;136:1585-1592.
[Article] [PubMed] [PMC]157. Anstee QM, Darlay R, Cockell S, Meroni M, Govaere O, Tiniakos D, et al.; EPoS Consortium Investigators. Genome-wide association study of non-alcoholic fatty liver and steatohepatitis in a histologically characterised cohort. J Hepatol 2020;73:505-515. Erratum in: J Hepatol 2021;74:1274-1275 Erratum in: J Hepatol 2023 Mar 13. doi: 10.1016/j.jhep.2023.02.028.158. Abul-Husn NS, Cheng X, Li AH, Xin Y, Schurmann C, Stevis P, et al. A protein-truncating HSD17B13 variant and protection from chronic liver disease. N Engl J Med 2018;378:1096-1106.
[Article] [PubMed] [PMC]159. Gerges SH, Wahdan SA, Elsherbiny DA, El-Demerdash E. Non-alcoholic fatty liver disease: An overview of risk factors, pathophysiological mechanisms, diagnostic procedures, and therapeutic interventions. Life Sci 2021;271:119220.
[Article] [PubMed]160. Satapathy SK, Sanyal AJ. Epidemiology and natural history of nonalcoholic fatty liver disease. Semin Liver Dis 2015;35:221-235.
[Article] [PubMed]161. Wang XJ, Malhi H. Nonalcoholic fatty liver disease. Ann Intern Med 2018;169:ITC65-ITC80.
[Article] [PubMed]162. Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology 1998;114:842-845.
[Article] [PubMed]163. Buzzetti E, Pinzani M, Tsochatzis EA. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016;65:1038-1048.
[Article] [PubMed]164. Fukuda N, Ontko JA. Interactions between fatty acid synthesis, oxidation, and esterification in the production of triglyceride-rich lipoproteins by the liver. J Lipid Res 1984;25:831-842.
[Article] [PubMed]165. Caligiuri A, Gentilini A, Marra F. Molecular pathogenesis of NASH. Int J Mol Sci 2016;17:1575.
[Article] [PubMed] [PMC]166. Takaki A, Kawai D, Yamamoto K. Molecular mechanisms and new treatment strategies for non-alcoholic steatohepatitis (NASH). Int J Mol Sci 2014;15:7352-7379.
[Article] [PubMed] [PMC]167. Alisi A, Carpino G, Oliveira FL, Panera N, Nobili V, Gaudio E. The role of tissue macrophage-mediated inflammation on NAFLD pathogenesis and its clinical implications. Mediators Inflamm 2017;2017:8162421.
[Article] [PubMed] [PMC]168. Deng CJ, Lo TH, Chan KY, Li X, Wu MY, Xiang Z, et al. Role of B lymphocytes in the pathogenesis of NAFLD: A 2022 update. Int J Mol Sci 2022;23:12376.
[Article] [PubMed] [PMC]169. Gehrke N, Schattenberg JM. Metabolic inflammation-A role for hepatic inflammatory pathways as drivers of comorbidities in nonalcoholic fatty liver disease? Gastroenterology 2020;158:1929-1947.e6.
[Article] [PubMed]170. Hassan K, Bhalla V, El Regal ME, A-Kader HH. Nonalcoholic fatty liver disease: a comprehensive review of a growing epidemic. World J Gastroenterol 2014;20:12082-12101.
[Article] [PubMed] [PMC]171. Loomba R, Friedman SL, Shulman GI. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 2021;184:2537-2564.
[Article] [PubMed]172. Peverill W, Powell LW, Skoien R. Evolving concepts in the pathogenesis of NASH: beyond steatosis and inflammation. Int J Mol Sci 2014;15:8591-8638.
[Article] [PubMed] [PMC]173. Hansen HH, Feigh M, Veidal SS, Rigbolt KT, Vrang N, et al. Mouse models of nonalcoholic steatohepatitis in preclinical drug development. Drug Discov Today 2017;22:1707-1718.
[Article] [PubMed]174. Lau JK, Zhang X, Yu J. Animal models of non-alcoholic fatty liver disease: current perspectives and recent advances. J Pathol 2017;241:36-44.
[Article] [PubMed] [PMC]175. Chen W, Chang B, Li L, Chan L. Patatin-like phospholipase domain-containing 3/adiponutrin deficiency in mice is not associated with fatty liver disease. Hepatology 2010;52:1134-1142. Erratum in: Hepatology 2010;52:2250.176. Tilson SG, Morell CM, Lenaerts AS, Park SB, Hu Z, Jenkins B, et al. Modeling PNPLA3-associated NAFLD using human-induced pluripotent stem cells. Hepatology 2021;74:2998-3017.
[Article] [PubMed]177. Kershaw EE, Hamm JK, Verhagen LA, Peroni O, Katic M, Flier JS. Adipose triglyceride lipase: function, regulation by insulin, and comparison with adiponutrin. Diabetes 2006;55:148-157.
[Article] [PubMed]178. Lake AC, Sun Y, Li JL, Kim JE, Johnson JW, Li D, et al. Expression, regulation, and triglyceride hydrolase activity of Adiponutrin family members. J Lipid Res 2005;46:2477-2487.
[Article] [PubMed]179. Wang SX, Yan JS, Chan YS. Advancements in MAFLD modeling with human cell and organoid models. Int J Mol Sci 2022;23:11850.
[Article] [PubMed] [PMC]180. Collin de l’Hortet A, Takeishi K, Guzman-Lepe J, Morita K, Achreja A, Popovic B, et al. Generation of human fatty livers using custom-engineered induced pluripotent stem cells with modifiable SIRT1 metabolism. Cell Metab 2019;30:385-401.e9.
[Article] [PubMed] [PMC]181. Kimura M, Iguchi T, Iwasawa K, Dunn A, Thompson WL, Yoneyama Y, et al. En masse organoid phenotyping informs metabolic-associated genetic susceptibility to NASH. Cell 2022;185:4216-4232.e16.
[Article] [PubMed]182. Wang S, Wang X, Tan Z, Su Y, Liu J, Chang M, et al. Human ESCderived expandable hepatic organoids enable therapeutic liver repopulation and pathophysiological modeling of alcoholic liver injury. Cell Res 2019;29:1009-1026.
[Article] [PubMed] [PMC]183. Singh S, Allen AM, Wang Z, Prokop LJ, Murad MH, Loomba R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and meta-analysis of paired-biopsy studies. Clin Gastroenterol Hepatol 2015;13:643-654.e1-9 quiz e39-40.
[Article] [PubMed] [PMC]184. Segovia-Miranda F, Morales-Navarrete H, Kücken M, Moser V, Seifert S, Repnik U, et al. Three-dimensional spatially resolved geometrical and functional models of human liver tissue reveal new aspects of NAFLD progression. Nat Med 2019;25:1885-1893.
[Article] [PubMed] [PMC]185. Chen Y, Gao WK, Shu YY, Ye J. Mechanisms of ductular reaction in non-alcoholic steatohepatitis. World J Gastroenterol 2022;28:2088-2099.
[Article] [PubMed] [PMC]186. Ben-Moshe S, Itzkovitz S. Spatial heterogeneity in the mammalian liver. Nat Rev Gastroenterol Hepatol 2019;16:395-410.
[Article] [PubMed]187. Purushotham A, Schug TT, Xu Q, Surapureddi S, Guo X, Li X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab 2009;9:327-338.
[Article] [PubMed] [PMC]188. Bhatia SN, Ingber DE. Microfluidic organs-on-chips. Nat Biotechnol 2014;32:760-772.
[Article] [PubMed]189. Wang Y, Wang H, Deng P, Tao T, Liu H, Wu S, et al. Modeling human nonalcoholic fatty liver disease (NAFLD) with an organoids-on-a-chip system. ACS Biomater Sci Eng 2020;6:5734-5743.
[Article] [PubMed]190. Ratziu V, Friedman SL. Why do so many NASH trials fail? Gastroenterology 2020 May 18;doi: 10.1053/j.gastro.2020.05.046.191. Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, et al.; NASH Clinical Research Network. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet 2015;385:956-965. Erratum in: Lancet 2015;385:946. Erratum in: Lancet 2016;387:1618.192. Pharm I. INT 747 (obeticholic acid) fails to achieve goals in Japanese study for non-alcoholic steatohepatitis (NASH). Amsterdam: The Netherlands: European Association for the Study of the Liver; 2015.193. Takebe T, Taniguchi H. Human iPSC-derived miniature organs: a tool for drug studies. Clin Pharmacol Ther 2014;96:310-313.
[Article] [PubMed]194. Yang K, Woodhead JL, Watkins PB, Howell BA, Brouwer KL. Systems pharmacology modeling predicts delayed presentation and species differences in bile acid-mediated troglitazone hepatotoxicity. Clin Pharmacol Ther 2014;96:589-598.
[Article] [PubMed] [PMC]195. Au SH, Chamberlain MD, Mahesh S, Sefton MV, Wheeler AR. Hepatic organoids for microfluidic drug screening. Lab Chip 2014;14:3290-3299.
[Article] [PubMed]