Programmed cell death in alcohol-associated liver disease
Article information

Abstract
Alcohol-associated liver disease (ALD), which ranges from mild disease to alcohol-associated hepatitis and cirrhosis, is the most prevalent type of chronic liver disease and a leading cause of morbidity and mortality worldwide. Accumulating evidence reveals that programmed cell death (PCD) plays a crucial role in progression of ALD involving crosstalk between hepatocytes and immune cells. Multiple pathways of PCD, including apoptosis, necroptosis, autophagy, pyroptosis and ferroptosis, are reported in ALD. Interestingly, PCD pathways are intimately linked and interdependent, making it difficult to therapeutically target a single pathway. This review clarifies the multiple types of PCD occurring in liver and focuses on crosstalk between hepatocytes and innate immune cells in ALD.
INTRODUCTION
Alcohol use is a major cause of preventable liver disease worldwide [1], and alcohol-associated liver disease (ALD), characterized by different stages including steatosis, fibrosis, cirrhosis and alcoholassociated hepatitis, is one of the most common causes of liver-related morbidity and mortality worldwide [2,3]. About 8–20% of chronic heavy drinkers will develop alcohol-related cirrhosis and of these patients, approximately 2% will develop hepatocellular carcinoma (HCC) [2].
Cell death is a fundamental biological process and plays a critical role in both health and disease. Cell death is important for growth and development, as well as maintaining a natural process for the turnover of aged cells, but it can also contribute to pathological conditions. Therefore, cell death is considered a “double-edge sword”, involved in both tissue homeostasis and pathogenesis. In ALD, programmed cell death (PCD), resulting, at least in part, from ethanol-induced oxidative stress and innate immune responses, is thought to play a central role in the progression of injury. ALD is a metabolic liver disease in which pathologic progression is largely driven by inflammatory responses [4,5]. Therefore, in this review, we briefly summarize the recent evidence on the role of multiple types of PCD and describe the critical role of perturbations in PCD in hepatocytes in the progression of ALD (Table 1).
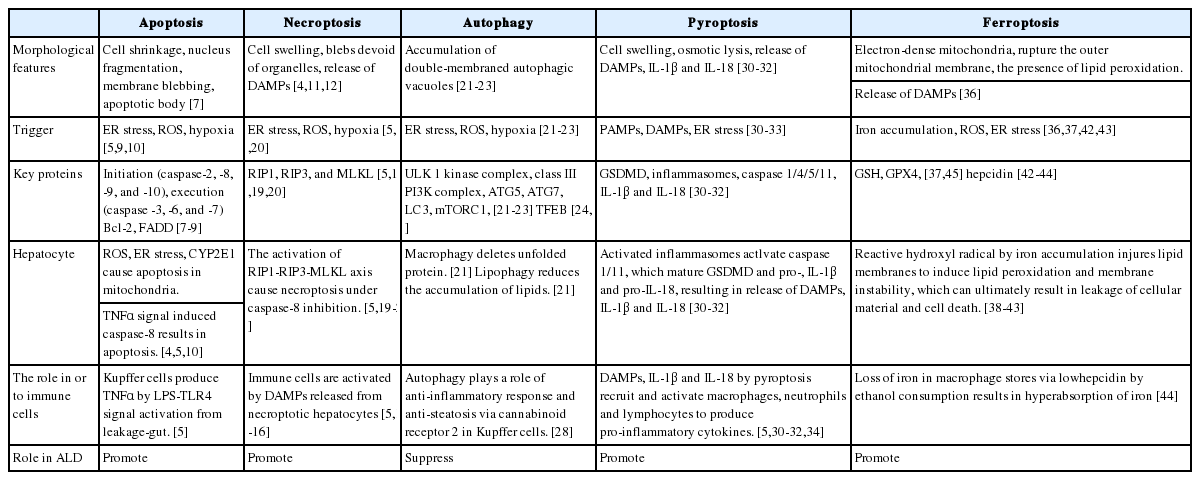
The characteristics of each programmed cell death pathway in ALD
PCD in ALD
Apoptosis
Apoptosis, a highly regulated mode of cell death, has been well studied since the pathway was first described in 1972 as a morphologically distinct type of cell death [6]. Apoptosis is a vital component of multiple homeostatic processes including normal cell turnover and the proper development and functioning of the immune system, and is a PCD mechanism characterized by caspase activation via the intrinsic- or extrinsic pathway [7]. Apoptosis results in cleavage of vital cellular proteins, chromatin condensation, nuclear fragmentation and membrane blebbing (Fig. 1). The initiation of apoptosis is dependent on the activation of the initiator- or the executioner caspases [7]. Once cell damage is detected, the initiator caspases (caspases-8/9) are activated from pro-caspases and activate the executioner caspases (caspases-3/6/7). The activation of the executioner caspases initiates a cascade of events that results in DNA fragmentation from activation of endonucleases, destruction of the nuclear proteins and cytoskeleton, and crosslinking of proteins. Apoptotic cells express ligands on their cell surface, which are recognized by phagocytic cells [7,8]. Apoptotic cells are therefore efficiently engulfed by surrounding macrophages, contributing to the non-inflammatory nature of this pathway of PCD.
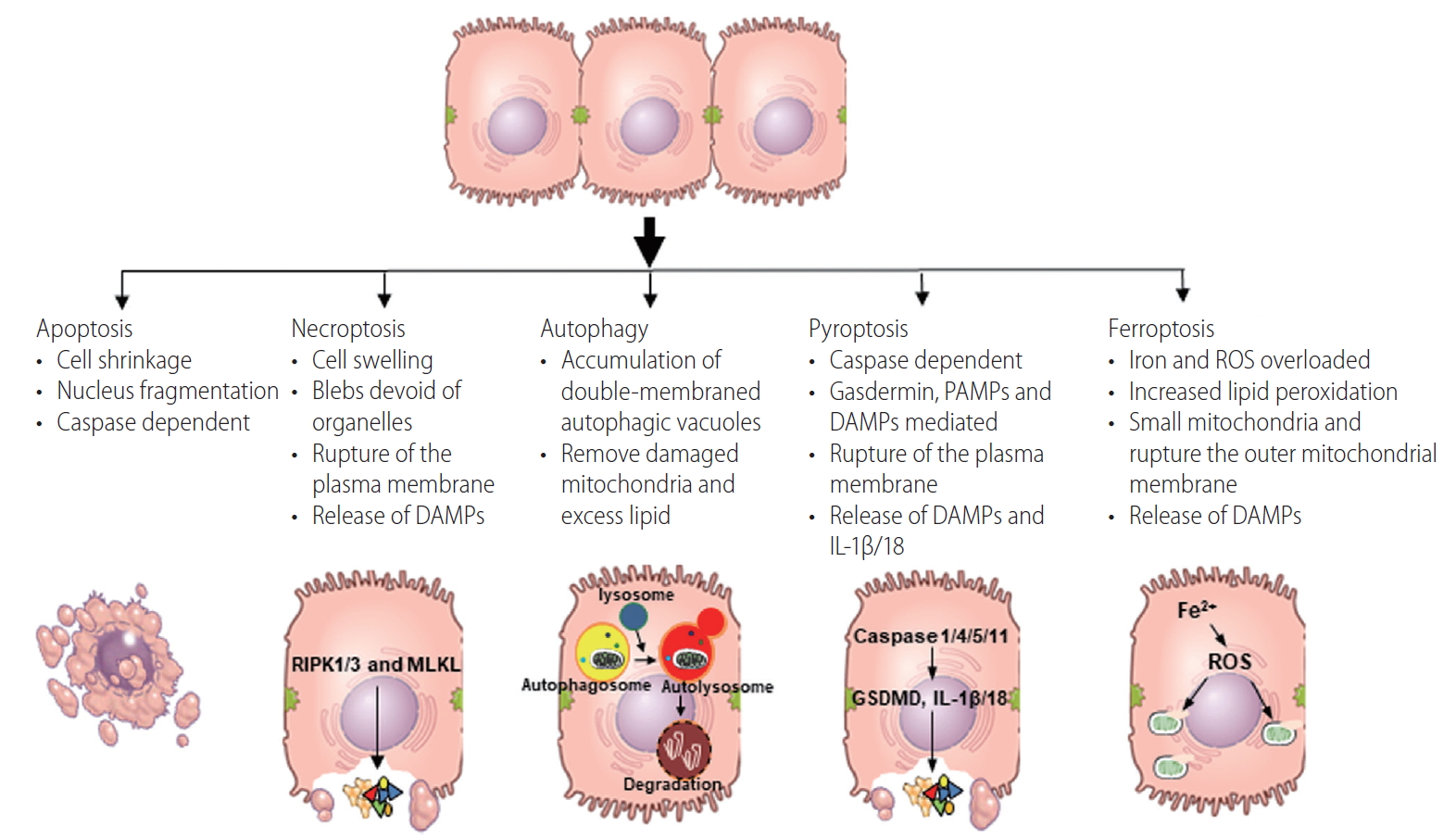
The features of each programmed cell death pathways. Figure shows typical features of each programmed cell death. DAMPs, damage-associated molecular patterns; PAMPs, pathogen-associated molecular patterns; IL, interleukin; ROS, reactive oxygen species.
Chronic alcohol exposure induces multiple forms of cellular stress, including oxidative stress, hypoxia and endoplasmic reticulum (ER)-stress, resulting in activation of both intrinsic (mitochondrial) and extrinsic (death-receptor mediated pathways including Fas/Fas Ligand, death receptor 4,5/tumor necrosis factor [TNF]-related apoptosis-inducing ligand and TNF receptor 1/TNFα) modes of apoptotic cell death [5,9,10]. Cellular stress is due both to ethanol metabolism and increased exposure to gut-derived pathogen associated molecular patterns (PAMPs) and endogenous danger associated molecular patterns (DAMPs). Ethanol metabolism results in both hypoxia and oxidative stress. Ethanol is oxidized by alcohol dehydrogenase, cytochrome P450 family 2, subfamily E, polypeptide 1 (CYP2E1) and catalase, resulting in the generation of reactive oxygen species (ROS). Ethanol metabolism also results in a rapid utilization of oxygen, contributing to a hypoxic environment and further ROS production in hepatocytes. Thus, alcohol metabolism can result in the formation of a variety of protein adducts, as well as impair proper protein folding in the ER, resulting in accumulation of misfolded proteins and ER stress, which induces apoptosis by activating caspase-12/4, c-Jun N-terminal kinase, and the interferon regulatory factor 3 signaling pathway [5].
In addition, ethanol promotes gut permeability allowing microbial lipopolysaccharides (LPS) into the portal circulation [5]. LPS in turn stimulates the production of pro-death mediators, such as TNFα, by resident hepatic macrophages and infiltrating monocytes via activation of toll-like receptor 4 [5]. These pro-death mediators can in turn activate the extrinsic pathway of apoptosis. Thus, ethanol promotes death receptor mediated hepatocyte cell death via activation of pro-inflammatory responses in innate immune cells [4].
Necroptosis
As early as 2005, a novel form of cell death exhibiting morphological characteristics of necrosis (Fig. 1), but, unlike necrosis, appeared to be tightly regulated. This mode of regulated cell death was identified and termed necroptosis [11]. Apopotosis and necroptosis share some of the same intracellular signaling mechanisms, with cell death shifting from apoptosis to necroptosis under conditions of low caspase-8 activity. Receptor interacting protein (RIP) 1, RIP3 and FAS-associated protein with death domain form a complex under these conditions, resulting in the association of RIP3 and mixed lineage kinase domain like pseudokinase (MLKL) and phosphorylation of MLKL [12]. Phosphorylated MLKL oligomerizes and then translocates to the plasma membrane where it forms pores to disrupt the plasma membrane integrity [12-15] and release of DAMPs such as mitochondrial DNA, highmobility group protein B1, and ATP [4,12]. Necroptotic cell death is immunogenic and promotes excessive inflammation and cell death by activation of innate immune cells or promoting other types of cell death, such as pyroptosis [14,16].
In ALD, understanding the role of necroptosis has been of interest, since inhibition of apoptosis in murine models of early ALD is not protective [17,18], suggesting that necroptosis might be important in ethanol-induced liver injury [19]. Indeed, RIP3 expression in liver is elevated in both murine models of ALD and patients with ALD. Chronic ethanol‐ and Gao-binge (acute on chronic) ethanol-induced injury were decreased in RIP3 knockout mice compared to wild-type, suggesting the importance of RIP3 in mediating ethanol‐induced liver injury and progression of ALD [19,20]. In contrast, high fat diet-induced liver injury, a model of non-alcoholic fatty liver and non-alcoholic steatohepatitis, was independent of RIP3, but instead dependent on MLKL. In the high-fat diet model, MLKL attenuated autophagic flux independently of RIP3 by inhibiting lysosomal fusion to autophagosomes [15]. However, no data is currently available on the potential role of MLKL in ethanol-induced liver injury.
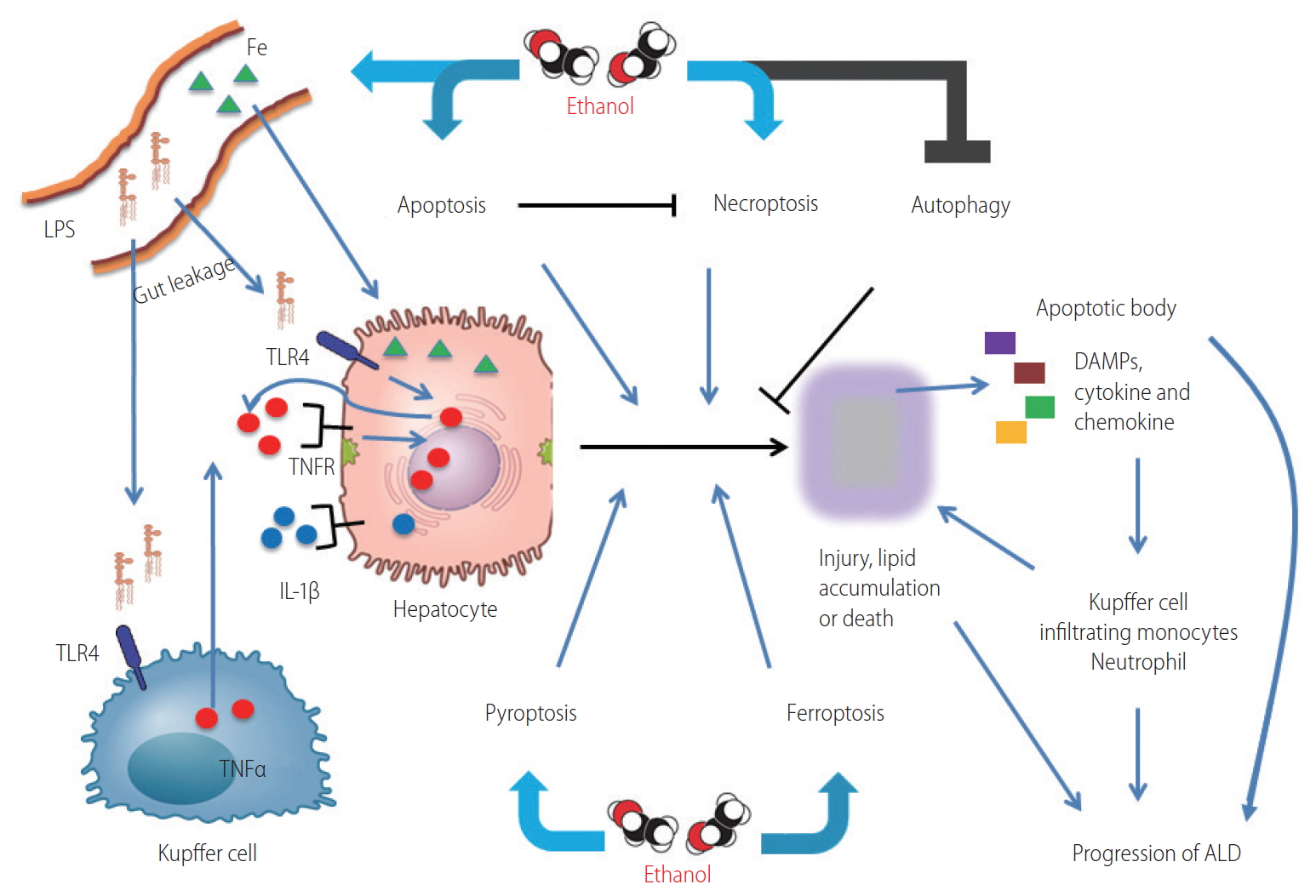
Overview of the role of programmed cell death pathways in ALD. Ethanol promotes ER stress, ROS production in hepatocytes and gut permeability. Gut microbiome derived LPS enters into liver via portal vein, stimulates hepatocytes and Kupffer cells, resulting in production of pro-inflammatory cytokines such as TNFα, IL-1β, IL-6, and MCP-1. These cytokines can induce death receptor signaling, resulting in apoptosis and necroptosis, the mechanism of cell death depends on caspase-8 activity. Necroptotic cells release DAMPs which activate pyroptosis, as well as ER stress driven by ethanol. Ethanol attenuates the protective functions of autophagy in response to chronic exposure and also promotes ferroptosis via ROS production and iron accumulation. Cumulatively, these changes result in lipid accumulation, increased production of pro-inflammatory cytokines and chemokines, hepatocellular injury and death, resulting in a spiraling progression of liver injury. LPS, lipopolysaccharides; TLR4, toll-like receptor 4; TNFα, tumor necrosis factor α; TNFR, TNF receptor; IL, interleukin; DAMPs, damage-associated molecular patterns; ALD, alcohol-associated liver disease; ER, endoplasmic reticulum; ROS, reactive oxygen species; MCP-1, monocyte chemoattractant protein-1.
Autophagy
Autophagy is considered a type II form of PCD [21], and is a regulated, catabolic process in which autophagosomes, double membrane vesicles, are formed de novo to engulf cytoplasmic content, which is then degraded upon fusion of the autophagosome with the lysosome. The molecular mechanism of autophagy, executed by the ULK1 kinase complex, the class III phosphatidylinositol 3-kinase complex, autophagy-related genes and microtubule-associated protein 1A/1B light chain 3, is well studied and reviewed [22-24]. In the growing cell, autophagy is maintained at low basal levels, where it serves as a quality control pathway, eliminating long-lived proteins and damaged organelles. Autophagy is also induced in response to cellular stress, such as nutrient starvation, growth factor withdrawal and energy depletion. Through degradation and recycling of cellular components, autophagy supplies a continual source of metabolic building blocks for cellular integrity. Thus, the main function of autophagy is to contribute to cellular renewal, allowing the lysosomal degradation of different components, including extracellular material and membrane proteins, cytosolic components and organelles to maintain cellular homeostasis [23,24]. Stress-induced autophagic responses prevent the accumulation of potentially pathogenic material in hepatocytes.
Autophagy is also associated with the progression of ALD [22]. In response to cellular stress such as increased production of highly reactive acetaldehyde, ROS, fatty acid ethyl esters and phosphatidylethanol generated during ethanol metabolism, hepatocytes induce multiple selective arms of autophagy, including parkin-mediated mitophagy and lipophagy, in order to remove damaged mitochondria and excess lipid, respectively [22]. Interestingly, autophagy is differentially regulated by acute vs chronic ethanol exposure [25]. Autophagy maintains hepatic homeostasis, protecting liver from alcohol-induced injury in response to acute ethanol challenge, serving to remove unfolded proteins and reduce the accumulation of lipids via lipophagy. However, after chronic alcohol exposure, autophagy is suppressed via multiple mechanisms [25]. First, chronic ethanol consumption inhibits mTOR activation, which leads to the suppression of initiation of autophagy [22]. Second, chronic ethanol increases lysosomal pH and impairs trafficking of lysosomal enzymes [26,27]. Chronic ethanol also inhibits TFEB expression, a transcription factor required for lysosomal biogenesis and autophagy [25,28]. Together, these changes inhibit autophagosome-lysosome fusion in hepatocytes [25]. In Kupffer cells, autophagy also plays a role of anti-inflammatory response and anti-steatosis via cannabinoid receptor 2 [29]. Thus, autophagy protects the liver from ethanol both in hepatocytes and Kupffer cells; however, over chronic periods of alcohol consumption, autophagy is suppressed, leading to accumulation of damaged organelles and dysplastic hepatocytes.
Pyroptosis
Pyroptosis, reported by Cookson and Brennan [30] in 2001, is primarily involved in pro-inflammatory events. Recent findings show that when inflammasomes are activated by PAMPs or DAMPs, inflammatory caspases (caspase 1, 4, and 5 in humans; caspase 1 and 11 in mice) are also activated. These caspases are required to mature pro-interleukin (IL)-1β/18 to IL-1β/18 and also to cleave gasdermin D to release its 31-kDa gasdermin-N domain that perforates the plasma membrane. The combination of these events results in the release of pro-inflammatory mediators, including IL-1β/18, as well as DAMPs [31-33]. These mediators then stimulate proinflammatory processes, including the recruitment and activation of neutrophils, macrophages and other immune cells [5].
Accumulating evidence shows that pyroptosis also plays a pivotal role in ALD pathogenesis. The NLR family pyrin domain containing 3 (NLRP3) inflammasome pathway is activated in hepatocytes in response to LPS-induced ER stress [34]. Importantly, NLRP3 deficiency ameliorates liver steatosis and injury by chronic ethanol. In addition, pyroptosis triggered by gut-derived PAMPs and by metabolic-derived DAMPs (uric acid and ATP) results in release of inflammasome-dependent cytokines from immune cells exposed to ethanol [35]. Thus, in ALD, pyroptosis is activated in both hepatocytes and liver immune cells, exacerbating inflammation in the liver via crosstalk between the different cell types. Heo et al. [36] demonstrated that ethanol promoted caspase-1-mediated pyroptosis through overexpression of thioredoxin-interacting protein, a member of the α-arrestin family, and this event is prevented by hepatocyte-specific delivery of micro RNA-148a. Taken together, these data indicate that pyroptosis contributes to stimulating and effectively sustaining the inflammatory cycle in ALD, although further studies on the cell specific contribution of NLRP3 inflammasomes to disease progression still requires further study.
Ferroptosis
Ferroptosis is a form of PCD characterized by the iron-dependent accumulation of lipid hydroperoxides to lethal levels. Ferroptosis was first described as a form of cell death in cancer by Stockwell and colleagues [37]. Morphologically, ferroptosis is characterized by cell volume shrinkage, electron-dense mitochondria, rupture of the outer mitochondrial membrane and the presence of lipid peroxidation [37]. Inactivation of glutathione peroxidase-4, which reduces lipid peroxides in plasma membranes, by relative or absolute depletion of glutathione (GSH) triggers ferroptosis by accumulation of ROS production from lipid peroxidation or Fenton reaction by excessive iron [38]. These reactive hydroxyl radicals damage lipid membranes, inducing lipid peroxidation and membrane instability, which can ultimately result in the leakage of cellular material and cell death.
Accumulating evidence suggests that ferroptosis is a novel type of PCD in liver diseases including ALD [39-42]. Hepatic iron accumulation is causally linked with chronic alcohol consumption, and it also contributes to hepatic inflammation and development of HCC in ALD [43,44]. Iron accumulates in response to ethanol, at least in part due to increased expression of hepcidin, a 25 amino-acid peptide, primarily expressed in hepatocytes, that is secreted from the liver in response to increased H2O2 concentrations in liver [44]. Hepcidin maintains iron homeostasis by inhibiting duodenal iron absorption, iron recycling from macrophages, and iron mobilization from hepatic stores by binding to the iron exporter, ferroportin [45]. Deletion of hepcidin in mice or hepcidin deficiency in humans results in severe hepatic iron overload, increased serum iron levels, and loss of iron in macrophage stores [43]. To date, little is known about ferroptosis in hepatocytes and immune cells in response to alcohol. However, one recent study found that overexpression of adipose-specific lipin-1 accelerated iron accumulation, caused lipid peroxidation, reduced GSH, and promoted ferroptotic liver damage in mice after ethanol administration [46]. Another study demonstrated that intestinal specific deficiency of Sirtuin 1, a class III histone deacetylase that plays a critical, but controversial, role in intestinal inflammation and development of colitis, protected mice from the chronic plus binge-induced liver damage by attenuating ferroptosis [40]. In the future, ethanol-induced crosstalk between hepatocytes and immune cells in the regulation of ferroptosis is an essential issue that needs to be clarified and understood in the context of ALD.
CONCLUSION AND FUTURE PERSPECTIVES
As reviewed here, PCD pathways have complex and interacting cell and tissue specific roles. However, recent evidence has revealed much in regard to their mechanisms of action and impact on disease progression. Understanding the mechanisms of PCD and crosstalk between hepatocytes and innate immune cells in ALD could provide a key to discovering innovative therapeutic targets or biomarkers that would contribute to improving the prognosis of patient’s with ALD. Therefore, it is essential to keep investigating PCD in ALD in order to elucidate the role of PCD in ALD.
Notes
Authors’ contribution
Drafting the article; TM, Critical revision of the article; LEN, and Final approval of the version to be published; TM and LEN
Conflicts of Interest: The authors have no conflicts of interests to disclose.
Acknowledgements
This work was supported in part by NIH grants; P50 AA024333, UO1 AA026938, R01 AA027456 and 1R01AA023722 (LEN), JSPS Overseas Research fellowship; 201960331 (TM).
Abbreviations
ALD
alcohol-associated liver disease
CYP2E1
cytochrome P450 family 2, subfamily E, polypeptide 1
DAMPs
damage-associated molecular patterns
ER
endoplasmic reticulum
GSH
glutathione
HCC
hepatocellular carcinoma
IL
interleukin
LPS
lipopolysaccharides
MLKL
mixed lineage kinase domain like pseudokinase
NLRP3
NLR family pyrin domain containing 3
PAMPs
pathogen-associated molecular patterns
PCD
programmed cell death
RIP
receptor interacting protein
ROS
reactive oxygen species
TNFα
tumor necrosis factor α