Current knowledge about biomarkers of acute kidney injury in liver cirrhosis
Article information

Abstract
Acute kidney injury (AKI) is common in advanced cirrhosis. Prerenal azotemia, hepatorenal syndrome, and acute tubular necrosis are the main causes of AKI in patients with cirrhosis. Evaluation of renal function and differentiation between functional and structural kidney injury are important issues in the management of cirrhosis. However, AKI in cirrhosis exists as a complex clinical spectrum rather than concrete clinical entity. Based on current evidence, changes in serum creatinine (Cr) levels remain the most appropriate standard for defining AKI in cirrhosis. However, serum Cr has a limited role in assessing renal function in this population. This review examines previous studies that investigated the ability of recent biomarkers for AKI in cirrhosis from the perspective of earlier and accurate diagnosis, classification of AKI phenotype, and prediction of clinical outcomes. Serum cystatin C and urine neutrophil gelatinase-associated lipocalin have been extensively studied in cirrhosis, and have facilitated improved diagnosis and prognosis prediction in patients with AKI. In addition, urine N-acetyl-β-D-glucosaminidase, interleukin 18, and kidney injury molecule 1 are other promising biomarkers for advanced cirrhosis. However, the clinical significance of these markers remains unclear because there are no cut-off values defining the normal range and differentiating phenotypes of AKI. In addition, AKI has been defined in terms of serum Cr, and renal biopsy—the gold standard—has not been carried out in most studies. Further discovery of innovate biomarkers and incorporation of various markers could improve the diagnosis and prognosis prediction of AKI, and will translate into meaningful improvements in patient outcomes.
INTRODUCTION
Renal dysfunction is a frequent and life-threatening complication of advanced cirrhosis [1-3]. It has been established that acute kidney injury (AKI) directly correlates with the prognosis of patients with cirrhosis [4-7]. Furthermore, a gradual increase in serum creatinine (Cr) was observed after the resolution of AKI, and the survival of patients with AKI was significantly reduced compared to that of patients without AKI [8]. AKI in cirrhosis is not a single disease entity; rather, it consists of complex pathophysiology. Hepatorenal syndrome (HRS) is a characteristic feature of advanced cirrhosis; however, it is not the only cause of AKI in these patients [9,10]. Accurate estimation of renal function is important for decision making including liver transplantation (LT). In addition, clearly differentiating the phenotype of AKI in cirrhosis is crucial, because the management of AKI is dictated by its cause.
One of the major limitations in managing patients with AKI and cirrhosis is insufficient ability of serum Cr to act as a kidney biomarker. Serum Cr usually overestimates the renal function in cirrhotic patients who have the high prevalence of sarcopenia [11]. In addition, because Cr is a marker reflecting kidney filtration, not injury, it is not suitable for differentiating the AKI phenotype. Overall, there is a distinct need for reliable biomarkers that can precisely assess renal function and differentiate the phenotypes of AKI in cirrhosis. Novel kidney biomarkers have been evaluated for AKI and cirrhosis to improve both diagnosis and clinical outcomes of the affected patients. This review examines the role of the current biomarkers in assessing renal function, predicting prognosis, and identifying the cause of kidney dysfunction.
DEFINITION AND PHENOTYPES OF AKI
AKI is a spectrum of clinical syndrome, including various precipitating factors that cause either direct injury to the kidney (structural injury) or cause acute dysfunction (functional injury). Risk, injury, failure, loss, end-stage kidney disease (RIFLE), and acute kidney injury network (AKIN) classifications based on changes in Cr and urine output were useful in predicting the prognosis of patients with liver cirrhosis [6,12-17]. In 2012, Kidney Disease: Improving Global Outcomes (KDIGO) proposed the new AKI definition; 1) increase in serum Cr by ≥0.3 mg/dL within 48 hours; or 2) increase in serum Cr to ≥1.5-fold of baseline, which is known or presumed to have occurred within the prior 7 days; or 3) urine volume <0.5 mL/kg/hour for 6 hours [18]. Several studies have validated this definition in hospitalized patients with cirrhosis [6,17,19,20]. However, in cirrhosis, urine volume could decrease without renal dysfunction or increase with the administration of diuretics [21]. Therefore, in 2015 International Club of Ascites consensus, AKI was defined using KDIGO serum Cr criteria, but urine volume was excluded from definition. A study with critically ill patients with cirrhosis showed that KDIGO criteria had higher predictability than RIFLE or AKIN criteria for assessing prognosis in these patients [22].
AKI phenotypes are classified as prerenal, intrarenal, and postrenal in the general population. Patients with cirrhosis characteristically develop a specific type of renal dysfunction, HRS [23]. Prerenal types represent approximately two-thirds of cases, whereas intrarenal AKI accounts for one-third of AKI cases, and postrenal AKI is rare in patients with cirrhosis [24]. Reversible AKI in cirrhosis is predominantly a prerenal injury, which resolves with volume administration and discontinuation of diuretics. In contrast, HRS is characterized by its nonresponsiveness to volume expansion [4]. HRS typically represents a continuum of disease, starting with functional changes, followed by structural changes due to prolonged ischemic injury. Intrinsic AKI involves acute tubular necrosis (ATN), acute interstitial nephritis, acute glomerular and vasculitic renal diseases. ATN is a condition that causes a lack of oxygen and blood flow to the kidneys, leading to damaged tubules. Proximal tubule is located in a vulnerable area exposed to hypoxic injury, and patchy areas of flattened tubular cells with enlarged lumens and apical vacuolization are the characteristic features of ATN. Renal tubules can also be injured with ischemia induced by prolonged hypoperfusion. Diagnostic approach of AKI in cirrhosis are presented in Figure 1. Accurately assessing renal function and differentiating the phenotypes of AKI are crucial in managing patients with cirrhosis. Although there have been many efforts, still there are many challenges and needs for relevant renal biomarkers for patients with cirrhosis.
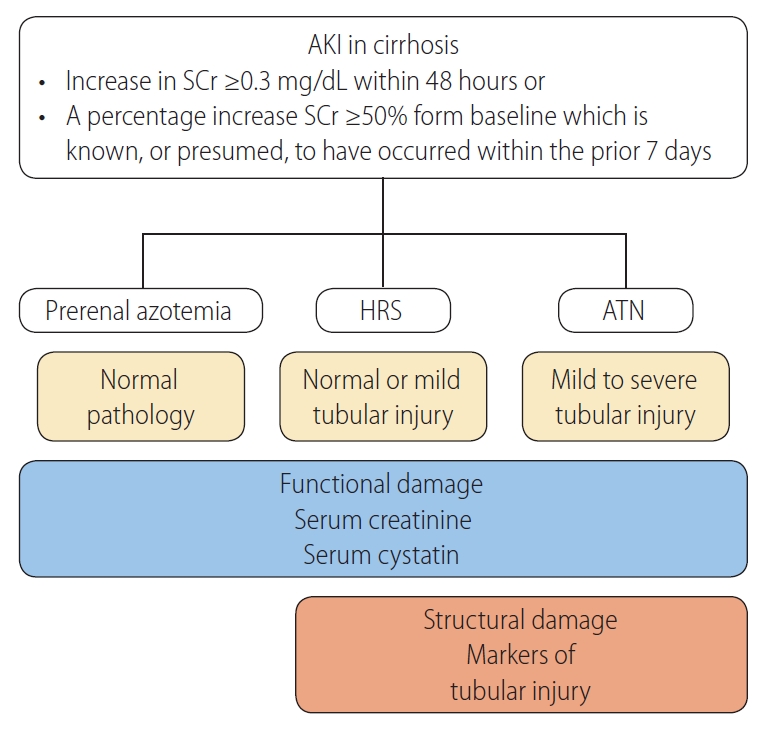
Diagnostic approach of AKI in cirrhosis. When AKI is diagnosed, volume administration and removal of precipitating factors are needed. Resolution of AKI is defined as a decrease in serum creatinine level to within 0.3 mg/dL of baseline value. When AKI persists after volume challenge, HRS and ATN could be the cause of AKI. HRS is the functional type of AKI, and are not expected to induce significant tubular damage, however, mild tubular injury could exist in HRS. In contrast, severe tubular lesions are characteristic feature of ATN. Functional biomarkers increase with severity of AKI, however, markers of structural damage appear in HRS and markedly increased in ATN. AKI, acute kidney injury; SCr, serum creatinine; HRS, hepatorenal syndrome; ATN, acute tubular necrosis.
PATHOPHYSIOLOGY OF HRS
The pathophysiology of HRS comprises several mechanisms, including hemodynamic alteration, kidney factors, and systemic inflammation (Fig. 2) [9,23]. Hypoperfusion plays a major role in the development of HRS. In early-stage cirrhosis, splanchnic vasodilatation results in a decrease in effective arterial volume, and it is balanced by hyperdynamic cardiac compensation to maintain adequate renal perfusion. In more advanced cirrhosis, the renin-angiotensin-aldosterone system, sympathetic nervous system, and arginine vasopressin are activated, leading to systemic vasoconstriction and sodium and water retention in the kidney [2,8]. At most of these stages, increased cardiac output fails to compensate, and this may contribute to a remarkable decrease in renal blood flow [25]. Impaired end-organ perfusion promotes functional kidney injury and a decrease in glomerular filtration rate (GFR), although there is no or minimal parenchymal kidney damage [26]. In addition, in response to changes in the systemic circulation, autoregulation of the renal blood flow by protective mediators could be impaired, further decreasing GFR [27].
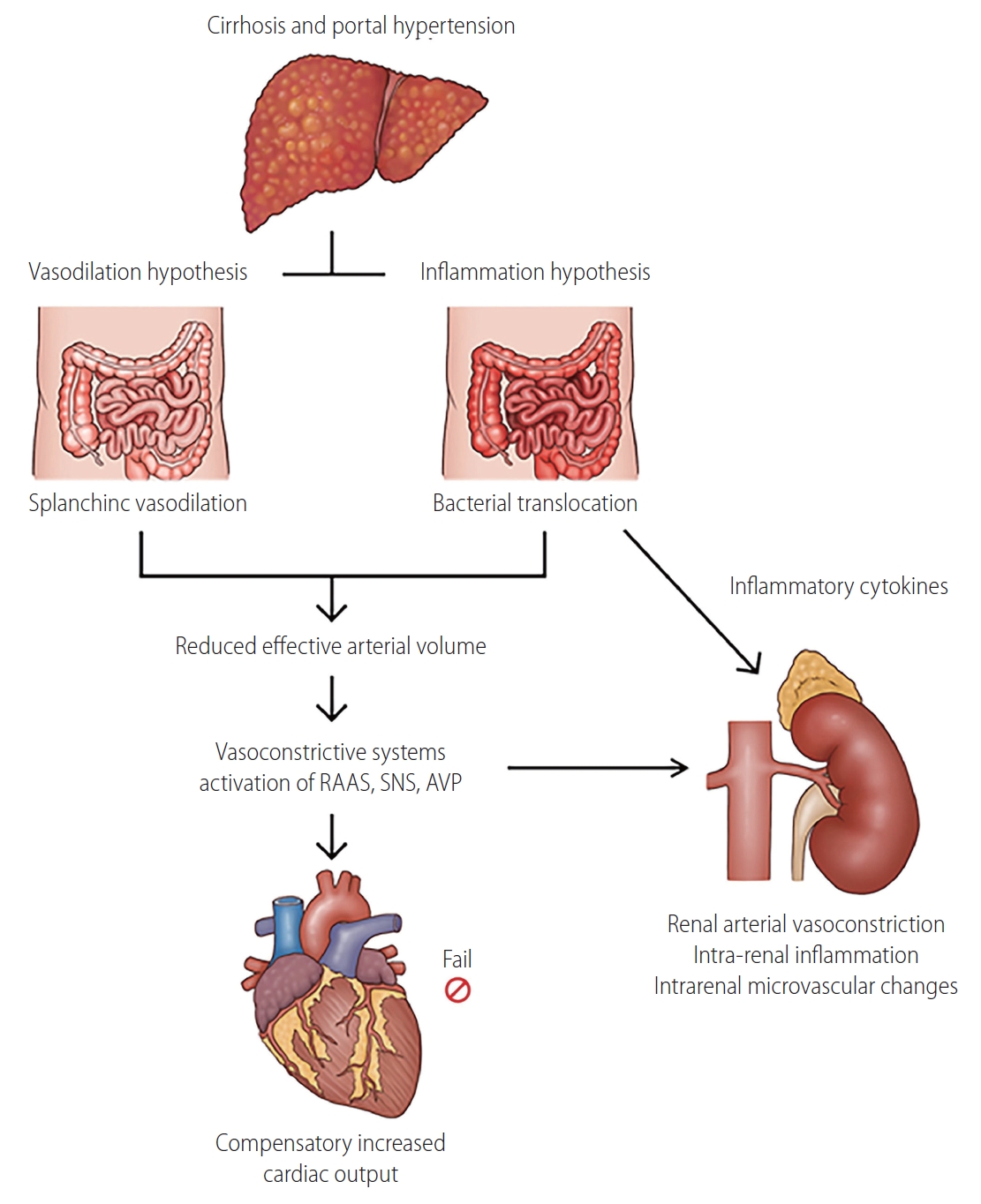
Pathophysiology of hepatorenal syndrome. In advanced cirrhosis, both vasodilation and systemic inflammation contribute to development of hepatorenal syndrome. Although increased cardiac output and activated systemic vasoconstrictors induce compensatory response, decreased effective arterial volume eventually leads to renal arterial vasoconstriction. Bacterial translocation induces intrarenal inflammation, resulting renal hypoperfusion and intrarenal microvascular changes. As a result, imbalance between pro- and post-glomerular resistance develops, and renal microcirculation affecting tubular and glomerular function is impaired, leading to decrease in glomerular filtration rate. RAAS, renin-angiotensin-aldosterone system; SNS, sympathetic nervous system; AVP, arginine vasopressin.
Recently, it has been recognized that HRS also involves systemic inflammation [28]. Gut permeability is easily disrupted in patients with cirrhosis, resulting in bacterial translocation. This induces the upregulation of systemic inflammatory mediators, leading to compromised extrahepatic organ perfusion, including the kidney [28-32]. In patients with bacterial translocation, pathogen-associated molecular patterns, such as endotoxins and bacterial DNA, activates monocytes, which leads to the release of pro-inflammatory cytokines such as tumor necrosis factor-alpha, interleukin (IL) 6, and IL-1 beta [33,34]. In addition, increased vasoactive mediators, such as nitric oxide, were observed in these patients. In previous studies, these cytokines were associated with impaired renal function in patients with cirrhosis, as well as in those with acute-on-chronic liver failure (ACLF) and acute liver failure [35,36].
It has been shown in both experimental and clinical studies that the renal tubular toll-like receptor 4 (TLR-4) is overexpressed following inflammatory insult in both experimental and clinical studies [37,38]. Upregulation of TLR-4 is related to the development of tubular cell damage, suggesting that structural changes in the kidney could also contribute to renal dysfunction in HRS. In an experimental study of rats with cirrhosis, tubular injury was observed after the administration of a sublethal dose of lipopolysaccharide, indicating that TLR-4 overexpression is likely a result of bacterial translocation [37]. In patients with sepsis-associated AKI, systemic inflammation-induced tubular cell injury is characterized by vacuolar degeneration, loss of polarity, loss of tight junctions, apoptosis, dedifferentiation and cell cycle arrest [39,40]. According to the results of recent studies, it has been suggested that structural damage could be combined in HRS, which is known to have predominant functional damage.
KIDNEY BIOMARKERS
With advances in proteomic platforms, several novel biomarkers of AKI have been discovered [41]. These biomarkers are used in preclinical models of drug development, for early recognition of nephrotoxicity [42]. In clinical studies, kidney biomarkers have been mainly assessed in intensive care unit patients and cardiac surgery populations, who have a high prevalence of AKI [43]. The ability of innovate kidney biomarkers has been investigated from several perspectives. Large prospective studies have demonstrated multiple biomarkers that can detect AKI earlier than could the previous markers [44,45]. Timely identification of AKI using these markers allows for the rapid management of and improved clinical outcomes in the affected populations. Several biomarkers are associated with the progression to a more severe stage of AKI, or long-term prognosis including the development of AKI, reversibility of renal impairment, and mortality [46-50]. Lastly, some biomarkers were useful in differentiating prerenal AKI from ATN, reducing unnecessary volume administration to those with fluid-unresponsive AKI [51,52]. Kidney markers in cirrhosis can be categorized into three groups: 1) markers of kidney dysfunction; 2) tubular injury markers increase with cellular damage, characterized by alteration in cell metabolism and expression of adhesion molecules; and 3) cell-cycle arrest marker detects cellular stress, which is potentially reversible and may or may not develop cell damage (Table 1) [53]. The summary of studies regarding novel kidney biomarkers are presented in Figure 3 and Table 2.

Characteristics of novel kidney biomarkers in cirrhosis
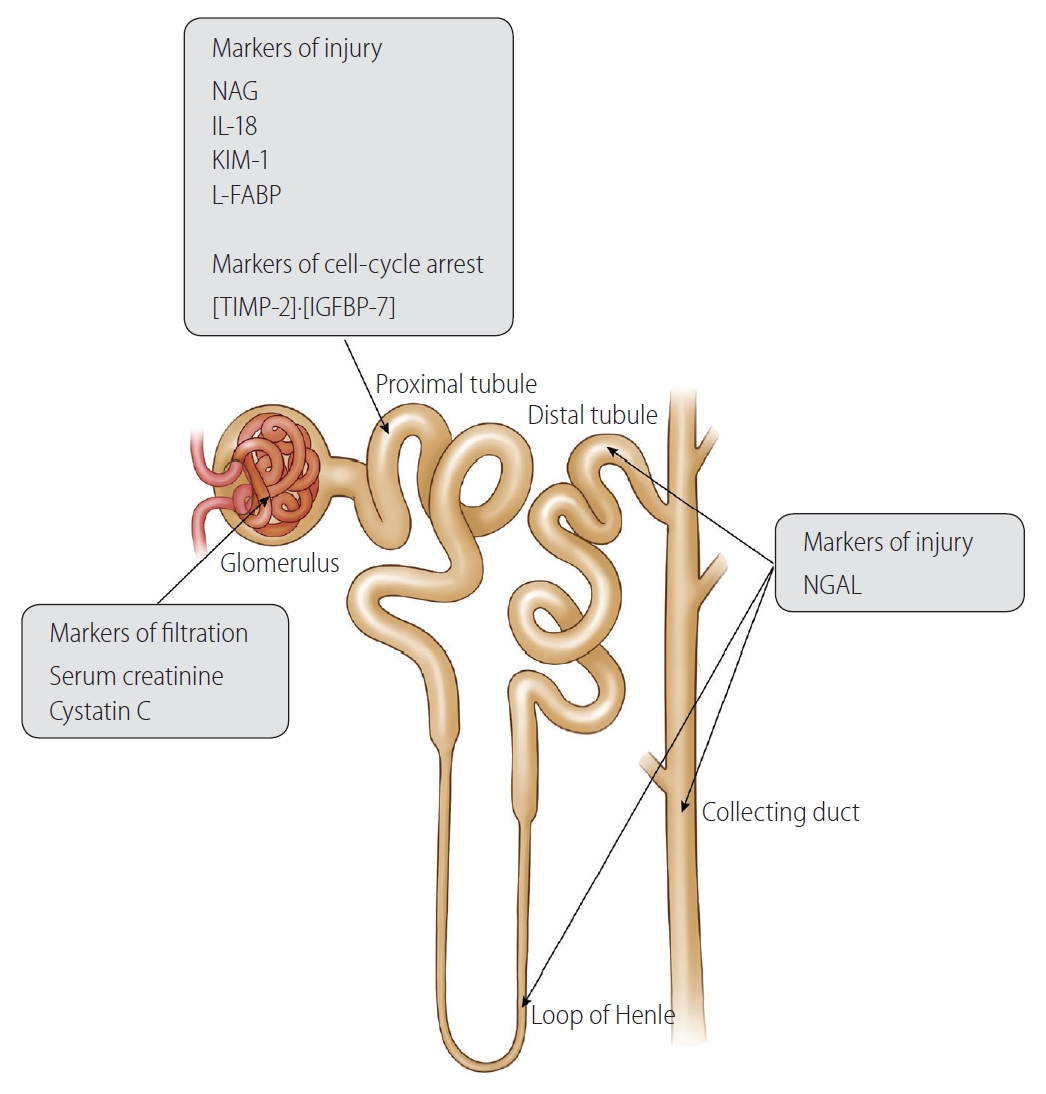
Overview of kidney biomarkers. NAG, N-acetyl-β-Dglucosaminidase; IL-18, interleukin 18; KIM-1, kidney injury molecule 1; L-FABP, liver-type fatty acid-binding protein; TIMP-2, tissue inhibitor of metalloproteinase-2; IGFBP7, insulin like growth factor binding protein 7; NGAL, neutrophil gelatinase-associated lipocalin.

Summary of studies regarding novel kidney biomarkers
BIOMARKERS FOR THE ASSESSMENT OF KIDNEY FUNCTION
Cr
Cr and Cr-based equations are the most widely used tools for estimating renal function in patients with liver cirrhosis. However, it is well established that serum Cr is not an accurate marker of renal dysfunction in cirrhosis [54-58]. Serum Cr levels vary with sex and age [57]. In some patients, the concentrations of serum Cr remain within normal limits, even in moderate to severe renal dysfunction, resulting in an overestimation of GFR [58-62]. Cr is produced in the liver and stored in the muscle, thus, its serum concentration is intimately related to liver disease and the amount of muscle mass [56,58]. In patients with hepatic dysfunction, the production of creatine, the precursor of serum Cr, is decreased, and tubular secretion of Cr is increased. Sarcopenia, which is prevalent in patients with advanced cirrhosis, also contributes to low serum Cr levels. Therefore, serum Cr level is insufficient for the detection of early-stage renal dysfunction in patients with cirrhosis, which could delay the timely management of kidney injury [60,63].
Inevitably, serum Cr level-derived equations (Cockcroft, modification of diet in renal disease [MDRD]-4, and Chronic Kidney Disease Epidemiology Collaboration equations) tend to overestimate GFR in patients with cirrhosis [11,64-66]. In addition, most serum Cr level-derived equations were developed in patients without cirrhosis, limiting its accuracy in cirrhosis [67-71]. In a large cohort study, MDRD-estimated GFR was overestimated in 47% of patients with liver cirrhosis, compared to the measured GFR, and female sex, advanced cirrhosis, and decreased skeletal muscle mass were independent predictors of overestimation [11]. Other studies have reported that Cr-based equations overestimate renal function by 30–50%, especially in patients with impaired liver function and low GFR [65-67,72].
Conflicting data have been reported regarding the association between Cr levels and the prognosis of cirrhosis. In some studies, a statistical significance between Cr and survival was observed, however, serum Cr levels were not significantly associated with mortality and or the development of HRS in other studies [60,72-75]. In addition, the observed mortality rate of patients with HRS was higher than that predicted by model for end-stage liver disease (MELD) or MELD sodium scores, which estimate renal function by serum Cr levels [76]. Currently, GFR is estimated using the Cr-based MDRD-6 equation in simultaneous liver-kidney transplant guidelines. Unfortunately, the measured GFR was underestimated when the measured GFR was greater than 30 mL/min/1.73 m2 in patients with cirrhosis, which could result in unnecessary simultaneous liver-kidney transplantation (SLKT) [77]. In contrast, the overestimation of the measured GFR could lead to increased mortality after LT [77,78].
Cystatin C
Serum cystatin C has been investigated to improve the detection of reduced GFR [79]. Cystatin C is a 13-kD endogenous cysteine proteinase inhibitor produced at a constant rate by all nucleated cells [80]. Cystatin C is eliminated mainly by glomerular filtration, and is almost completely reabsorbed and catabolized in tubules [81]. Serum cystatin C is not affected by sex, hepatic function, or muscle mass [55,82-85]. The kinetics of serum cystatin C are similar to those of Cr, with the serum concentration increasing 12–24 hours after the onset of AKI [86].
The accuracy of cystatin C-based estimated GFR was compared with that of Cr-based estimated GFR. In a study of 779 patients with cirrhosis, cystatin C-based estimated GFR showed a better correlation with measured GFR than the MDRD-estimated GFR [11]. Other studies demonstrated that Cr-cystatin C combined GFR equations were more accurate than Cr-based equations in predicting measured GFR in patients with cirrhosis, especially when the GFR was lower than 60 mL/min/1.73 m2 in both, the patients with and without ascites [87,88]. In patients with normal range of serum Cr levels, cystatin C was the only independent predictor for the measured GFR [55].
Cystatin C was more accurate than Cr for predicting outcomes of cirrhosis. In patients with cirrhosis, baseline cystatin C-based GFR (cut-off <55 mL/min/1.73 m2) was useful in the early recognition of AKI. In a prospective study of patients with cirrhosis, cystatin C was better at predicting overall survival and the incidence of AKI than MDRD-estimated GFR [11]. Other prospective study of 531 patients with cirrhosis, cystatin C level but not serum Cr was an independent predictor of AKI development, an MELD-cystatin score accurately predicted the AKI development and mortality [89]. In patients with cirrhotic ascites, cystatin C was an independent predictor of mortality and the development of HRS, while Cr was not [75]. Several studies have suggested that changes in cystatin C levels are associated with the development of AKI, however, these are insufficient for supporting the role of change in cystatin C level in defining AKI in cirrhosis [90,91].
BIOMARKERS FOR DEFINING THE PHENOTYPE OF AKI
Identification of AKI phenotype is crucial for establishing a treatment strategy and improving clinical outcomes. Because a clear objective test for HRS is absent, 48 hours of empirical volume expansion is required for both a diagnostic and therapeutic challenges. AKI caused by HRS and ATN persists after volume administration and requires appropriate management for each entity. However, differentiating HRS from ATN is challenging. The major treatment for HRS is the administration of vasoconstrictors, which has been shown to improves the renal function in approximately 50% of the patients with HRS [92-94]. However, short-term mortality of HRS remains very high, up to 90%, and LT is the only treatment that improves the long-term survival of patients with HRS [17,92,95]. In contrast, treatment of the underlying cause is the main therapeutic strategy for ATN. A fluid-first treatment may delay the initiation of vasoconstrictors in patients with HRS, or induce volume overload in those with ATN, who are unlikely to respond, resulting in a poor clinical outcome [96,97].
The level or change in serum Cr cannot differentiate between functional and structural renal impairment [98]. Fractional excretion of filtered sodium (FENa) is used to differentiate the phenotypes of AKI, characterized by FENa less than 1% in prerenal azotemia. However, low FENa can be observed in AKI subtypes other than the prerenal type because of activated water and sodium retention [23,99]. In addition, FENa can be increased by diuretics, glycosuria or chronic adaptation [100]. Proteinuria of over 1–2 g/day suggests a glomerular injury; however, in patients with cirrhosis, serum protein concentration is decreased by impaired hepatic function and poor nutrition, leading to a lower amount of proteinuria [18]. Glomerular injury can be suspected in patients with proteinuria of less than 0.5 g/day when their serum protein level is low. In addition, a poor correlation between conventional biomarkers and kidney histology was observed in a previous study [101]. Because of the disappointing performance of the previous markers in assisting with the differentiation of HRS from ATN and other kidney diseases, there is a great need to identify markers specific to parenchymal kidney insult. Differentiating functional changes from structural changes could help differentiate between patients with a reversible disease and those with irreversible changes.
Tubular injury biomarkers
Tubular injury biomarkers have been shown to have the potential to differentiate HRS from ATN in patients with cirrhosis. Hypoxia leads to proximal tubule dysfunction, resulting in increased excretion of low-molecular-weight proteins into urine or serum. Since they are expressed only hours after kidney injury, they have suggested as the earliest markers of AKI [102-104]. However, there are insufficient data to support their role as early detection markers of AKI in cirrhosis.
The levels of urinary tubular markers are different between AKI phenotypes, with the lowest levels observed in prerenal AKI, modest levels exhibited in HRS, and significantly higher levels found in ATN [105-110]. The potential tubular injury biomarkers in cirrhosis are 1) neutrophil gelatinase-associated lipocalin (NGAL), 2) N-acetyl-β-D-glucosaminidase (NAG), 3) IL-18, 4) kidney injury molecule 1 (KIM-1), and 5) liver-type fatty acid-binding protein (L-FABP).
NGAL is a low-molecular-weight protein (25 kDa) produced by thick ascending and collecting ductal cells of kidney and cells of liver, lung, and gastrointestinal tract [111]. In animal models, NGAL is highly expressed in the kidney and released into the urine after ischemic or nephrotoxic insults. Following ischemic insult, urine NGAL concentration increases within 2 hours [111]. In addition, NGAL plays a role in the innate immune system through iron sequestration [112,113]. Although both serum and urine NGAL can be measured, the serum NGAL levels have not been widely studied.
NAG originates from the lysosome of proximal tubules; therefore, increased urinary NAG levels represent proximal tubule injury with loss of lysosomal integrity [114]. Unlike NGAL, NAG has a large molecular weight (140 kDa) and is not filtered by the glomerulus; therefore, its high urine levels are unlikely to originate from extrarenal sources [115]. In several studies, urine NAG was shown to be a sensitive marker of tubular injury [114,116-118].
IL-18 is a pro-inflammatory cytokine that is highly expressed in proximal tubule. IL-18 is released into the urine after ischemic injury to the proximal tubule, and its level is not confounded by urinary infections, nephrotoxic AKI, and chronic kidney disease (CKD) [108,119-121]. In human studies, urinary IL-18 was useful in predicting the development of AKI and other clinical outcomes [122,123].
KIM-1 is a transmembrane protein that is expressed in proximal tubule and is upregulated in response to ischemic kidney injury. Because urine KIM-1 is less affected by extrarenal confounders, its role in differentiation of ATN from non-tubular kidney injury was observed [124-126].
-FABP is a free fatty acid transporter, which is overexpressed in the proximal tubule, and which transports free fatty acids to the mitochondria or peroxisomes [127]. In experimental studies, L-FABP has been observed to be upregulated and released into the urine in the presence of tubulointerstitial damage [128]. Human studies have shown that urinary L-FABP could predict the AKI or sepsis complicated by AKI, and its level could be affected by infection or liver disease [129,130]. In addition, urinary L-FABP levels are also increased in patients with CKD, suggesting its role as an antioxidant and renoprotective substance [131].
The role of tubular markers in the diagnosis of AKI
Several studies have shown that both serum and urine NGAL may be useful for the early diagnosis of AKI [45,132-134]. Thereafter, diagnostic value of NGAL was investigated in patients with liver cirrhosis, demonstrating a higher urinary NGAL level in patients with AKI than in those without AKI [135]. However, urinary NGAL levels also increase during other conditions, such as inflammatory disease, urinary tract infection, or CKD, limiting the diagnostic performance of NGAL [136,137]. In addition, it was shown that liver synthesis of NGAL increases during sepsis; therefore, NGAL level could be confounded [138,139]. In a recent multicenter prospective study of 328 patients with decompensated cirrhosis, urine NGAL levels were comparable between patients with and without AKI [140]. Furthermore, when multiple kidney biomarkers were analyzed simultaneously, increased serum cystatin C and urine NAG levels at baseline were significant predictors of AKI development, while serum Cr and urine NGAL levels were not. When patients were divided in to three groups according to the serum cystatin C and urine NAG levels, the hazard ratios for development of AKI were 3.010 and 6.512 in the other groups, compared to low-risk group. To date, the clinical implications of urine NAG have not been fully investigated; therefore, further validation studies in patients at a higher risk of AKI are needed.
Differentiation of AKI phenotype with tubular markers
Recently, NGAL and IL-18 helped identify the cause of AKI in patients with cirrhosis [105,106,135,141]. It has been suggested that urine NGAL is significantly higher in patients with ATN than those with other causes, including HRS and prerenal AKI [105,135]. In a study of 112 patients with various phenotypes of AKI, urine NGAL showed the highest diagnostic performance in differentiating ATN from other causes (area under curve [AUC] of 0.79), compared to IL-18, KIM-1, and L-FABP [105]. A meta-analysis suggested that urine NGAL and IL-18 could discriminate ATN from other AKI phenotypes with AUCs of 0.89 and 0.88, respectively [142]. The role of KIM-1 in patients with cirrhosis and AKI has been investigated in few studies, and increased urine KIM-1 levels were observed in patients with ATN compared to those with other causes [105]. However, in some studies, significant overlaps in urine NGAL, IL-18, and KIM-1 levels were observed between patients with HRS and those with ATN [106,135]. In addition, various previous studies have defined ATN and other causes of AKI with clinical features, without histological confirmation, which could lead to a misclassification of the AKI phenotype. None of these biomarkers were specific to any part of the nephron. Many patients with AKI present oliguria or anuria, and urine collection from these patients may be impractical, precluding the measurement of urinary biomarkers. Therefore, the performance of these markers in differentiating AKI phenotypes in cirrhosis should be interpreted with caution.
Patient outcomes and tubular injury markers
Conflicting results have been reported regarding the association between tubular markers and the risk of mortality [105,106,119,139-141]. In a meta-analysis of five studies, increased urine levels of IL-18 and NGAL identified patients at a higher risk of short-term mortality with an AUC of 0.76 [142]. In two studies investigating the performance of multiple biomarkers, urine NGAL levels showed the best predictive performance in predicting mortality, compared with the urine IL-18, KIM-1, and L-FABP levels and serum cystatin C level [105,108]. However, two recent prospective studies from South Korea, including patients with decompensated cirrhosis, showed that urine NGAL level was not a significant predictor of mortality; rather, the cystatin C or MELD-cystatin C score were associated with survival outcomes [123,140]. These discrepancies could be explained by the differences in the clinical characteristics of the patient population. Unlike previous studies that included a wide range of cirrhotic patients, the latter two studies only included patients with decompensated cirrhosis, to investigate the ability of the biomarkers appropriately in patients with a high prevalence of AKI. Another concern is that an increase in these biomarkers is not fully derived from kidneys. As mentioned above, infection or liver disease, the other critical factor of increased mortality, also increase the levels of renal biomarkers, especially NGAL and L-FABP. Therefore, it is not clear whether the mortality in the previous studies was driven by AKI itself.
Predicting kidney outcomes in patients with AKI and cirrhosis is also of great concern. In a prospective study of European AKI cohort in which terlipressin was applied as a treatment algorithm, urine NGAL and IL-18 levels were measured serially [107]. It was demonstrated that a high urinary NGAL level on hospital day 3, measured after 48 hours of volume expansion, was the most significant predictor of AKI progression; however, it was not related to the response to terlipressin. In another study, higher urinary NGAL, IL-18, KIM-1, and L-FABP levels were independently associated with the progression of AKI, with IL-18 performing best [105]. However, there are insufficient data to draw firm conclusions about their role in predicting kidney outcomes in patients with cirrhosis and AKI.
Biomarker for cell-cycle arrest
Tissue inhibitor of metalloproteinase-2 and insulin like growth factor binding protein 7 ([TIMP-2]·[IGFBP7]) is a recently discovered cell-cycle arrest protein expressed in renal tubular cells during cellular stress or injury. It was discovered through an unbiased proteomic screening in human AKI populations. TIMP-2 inhibits the activity of matrix metalloproteinase, affecting the regulation of the cell cycle, and binds to α3β1 integrin on the surface of endothelial cells, inhibiting endothelial cell proliferation and angiogenesis [143,144]. IGFBP7 is a secreted protein that regulates the bioavailability of IGFs through direct binding [145]. The role of [TIMP-2]·[IGFBP7] as a biomarker in AKI patients has been proposed because G1 cell cycle arrest is a known consequence of AKI [146]. Nephrocheck®, which detects TIMP-2 and IGFBP7, has been approved by the U.S. Food and Drug Administration as a potential biomarker for predicting clinical outcomes in patients with AKI. Although it was useful in predicting moderate to severe AKI within 12 hours and clinical outcomes in critically ill patients, few studies described an unclear role of [TIMP-2]·[IGFBP7] in patients with cirrhosis [123,147].
RENAL BIOMARKERS IN ACLF
In previous studies with patients with ACLF defined by both the Asia Pacific Association of the Study of Liver and the joint European Association of Study of Liver/American Association of Study of Liver Disease criteria, the prevalence of kidney dysfunction was reported as 13.2–51.0% [35,148-150]. Therefore, there is a distinct need for reliable renal biomarkers in patients with ACLF. Patients with ACLF have marked hyperbilirubinemia, resulting in low serum Cr level compared to the extent of residual renal function. A study showed that serum cystatin C level was useful in predicting renal impairment in patients with hepatitis-B virus related ACLF patients [151]. Urine NGAL was shown as a useful biomarker for the diagnosis and prognosis prediction in patients with ACLF [152]. In a study of patients with ACLF, urine NGAL and plasma soluble CD163 were independent predictors for 28-day mortality [153]. In the diagnosis of AKI phenotype in ACLF, differentiating bacterial infection-associated HRS from ATN, which is a continuum, is a challenging issue. However, data regarding the role of novel renal biomarkers for the differentiation of AKI phenotype in ACLF patients are lacking [154].
CONCLUSIONS
AKI has important prognostic implications in liver cirrhosis. It has been suggested that some consequences of AKI may not be fully recovered; rather, prolonged kidney vasoconstriction could induce tubular interstitial fibrosis and increase the risk of further AKI [155,156]. Unlike other populations, cirrhosis is the only disease that has a specific therapy for AKI, terlipressin, which is approved for the treatment of HRS in various regions. Therefore, the accurate evaluation of renal function and the determination of the AKI phenotype are important issues in the management of patients with AKI. Due to the complex pathophysiology and interaction between the liver and kidney, novel renal biomarkers have been adopted and evaluated. Although various markers have been shown to improve both diagnosis and prognosis prediction in patients with cirrhosis, a single biomarker is insufficient for having an impact on clinical practice. In addition, renal biopsy, which is essential to identify the etiology of AKI, was not performed in a majority of the relevant previous studies. And the normal range of novel markers in cirrhosis has not been defined, limiting their clinical use. Therefore, other novel biomarkers still need to be explored, and the incorporation of various biomarkers should be evaluated in future studies. The availability of these markers in cirrhosis will also help to further develop and improve the performance of certain current treatment strategies, including the administration of terlipressin and SLKT.
Notes
Authors’ contribution
Conception and design: Y.S. Seo; Writing, review, and/or revision of the manuscript: H.A. Lee and Y.S. Seo; Administrative, technical, or material support: Y.S. Seo; Study supervision: Y.S. Seo.
Conflicts of Interest: The authors have no conflicts to disclose.
Abbreviations
ACLF
acute on chronic liver failure
AKI
acute kidney injury
AKIN
acute kidney injury network
ATN
acute tubular necrosis
AUC
area under curve
CKD
chronic kidney disease
Cr
creatinine
FENa
fractional excretion of filtered sodium
GFR
glomerular filtration rate
HRS
hepatorenal syndrome
IGFBP7
insulin like growth factor binding protein 7
IL
interleukin
KDIGO
Kidney Disease: Improving Global Outcomes
KIM-1
kidney injury molecule 1
L-FABP
liver-type fatty acid-binding protein
LT
liver transplantation
MDRD
modification of diet in renal disease
MELD
model for end-stage liver disease
NAG
N-acetyl-β-D-glucosaminidase
NGAL
neutrophil gelatinase-associated lipocalin
RIFLE
risk, injury, failure, loss, end-stage kidney disease
SLKT
simultaneous liver-kidney transplantation
TIMP-2
tissue inhibitor of metalloproteinase-2
TLR-4
toll-like receptor 4