Prognostic value of ultra-low-pass whole-genome sequencing of circulating tumor DNA in hepatocellular carcinoma under systemic treatment
Article information

Abstract
Background/Aims
New prognostic markers are needed to identify patients with hepatocellular carcinoma (HCC) who carry a worse prognosis. Ultra-low-pass whole-genome sequencing (ULP-WGS) (≤0.5× coverage) of cell-free DNA (cfDNA) has emerged as a low-cost promising tool to assess both circulating tumor DNA (ctDNA) fraction and large structural genomic alterations. Here, we studied the performance of ULP-WGS of plasma cfDNA to infer prognosis in patients with HCC.
Methods
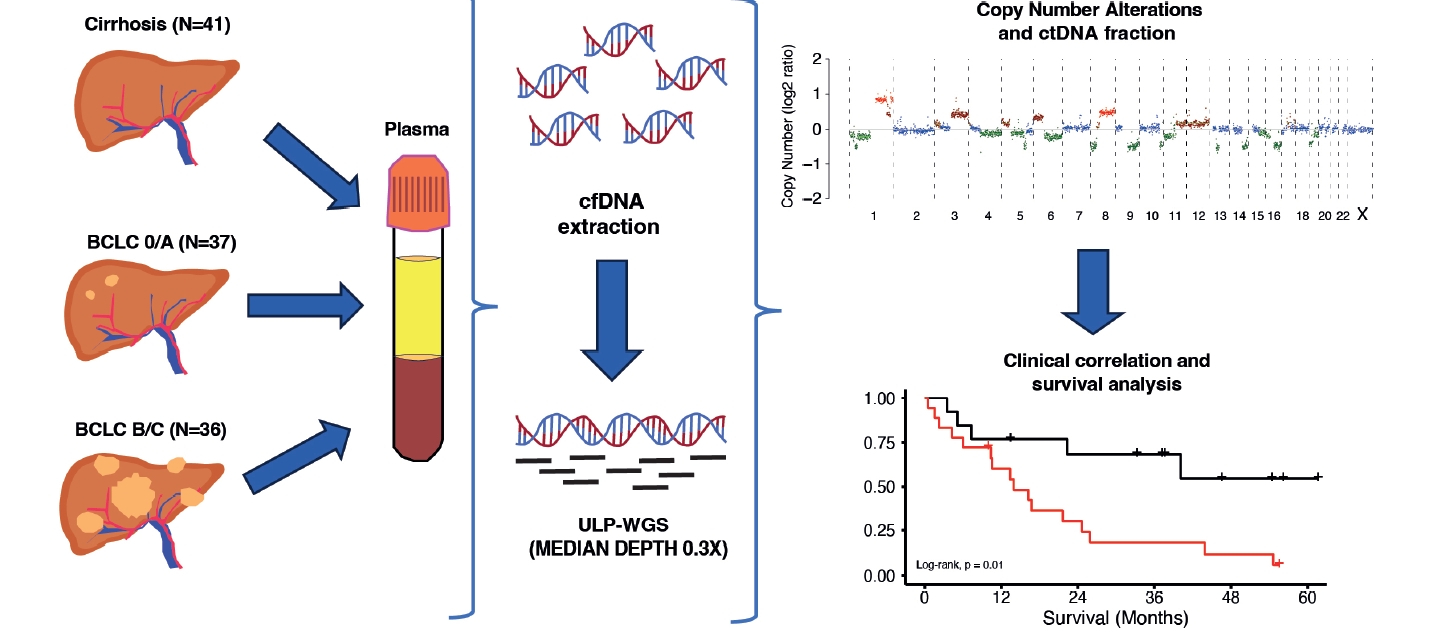
Plasma samples were obtained from patients with HCC prior to surgery, locoregional or systemic therapy, and were analyzed by ULP-WGS of cfDNA to an average genome-wide fold coverage of 0.3x. ctDNA and copy number alterations (CNA) were estimated using the software package ichorCNA.
Results
Samples were obtained from 73 HCC patients at different BCLC stages (BCLC 0/A: n=37, 50.7%; BCLC B/C: n=36, 49.3%). ctDNA was detected in 18 out of 31 patients who received systemic treatment. Patients with detectable ctDNA showed significantly worse overall survival (median, 13.96 months vs not reached). ctDNA remained an independent predictor of prognosis after adjustment by clinical-pathologic features and type of systemic treatment (hazard ratio 7.69; 95%, CI 2.09–28.27). Among ctDNA-positive patients under systemic treatments, the loss of large genomic regions in 5q and 16q arms was associated with worse prognosis after multivariate analysis.
Conclusions
ULP-WGS of cfDNA provides clinically relevant information about the tumor biology. The presence of ctDNA and the loss of 5q and 16q arms in ctDNA-positive patients are independent predictors of worse prognosis in patients with advanced HCC receiving systemic therapy.
Graphical Abstract
INTRODUCTION
Hepatocellular carcinoma (HCC) is one of the most common cancers worldwide with an unfavorable prognosis, particularly for patients with advanced disease. HCC most often develops on a cirrhotic liver, primarily due to viral hepatitis, alcohol-related liver disease, or metabolic liver disease [1,2]. According to the Barcelona Clinic Liver Cancer (BCLC) classification, patients with very early and early stages (BCLC 0-A) are the best candidates for ablative therapies, such as radiofrequency ablation, liver resection, or liver transplantation [3]. Intermediate stage HCC (multinodular liver-only disease, BCLC-B) is usually addressed with locoregional treatments when tumor burden is low, whereas advanced stages, such as BCLC-B with high tumor burden, or those tumors with vascular invasion or extrahepatic spread (BCLC-C) deserve systemic treatment [3]. However, within a specific tumor stage, patients may exhibit different prognosis or be treated with different therapies [4,5].
In this scenario of multiple therapeutic options, clinical, laboratory and pathological features may help inform clinical decision-making by providing important prognostic information. Microvascular invasion or satellite nodules, for instance, may establish an indication for ab initio liver transplantation [6] or adjuvant immunotherapy [7]. Serum bilirubin and albumin, or composite scores that incorporate them, such as Child-Pugh or albumin-bilirubin grade, may define the indication of systemic or locoregional therapies [3]. Among non-invasive tumor-derived biomarkers, only serum alpha-fetoprotein (AFP) has enough high-level supporting evidence to be used in clinical practice. High AFP levels may contraindicate liver transplantation [8] or define the indication of Ramucirumab, a VEGFR-2 inhibitor [9]. However, AFP has limited value, as only a minority of patients have increased levels (10% of patients at early stages have AFP >400 ng/dL scaling up to 40% in advanced stages) [10,11]. On the other hand, early changes in AFP may predict the benefit of systemic agents like Ramucirumab [9] or the combination of Atezolizumab plus Bevacizumab [12].
Thus, there is a clear need for novel biomarkers that may help in predicting the prognosis and monitoring treatment response, thereby guiding a more personalized therapy. Liquid biopsy using peripheral blood content can provide information about the primary tumor in a non-invasive manner. This is because tumors shed different elements into the blood, including tumor nucleic acids (DNA and RNAs), circulating tumor cell, and exosomes, which carry with them the molecular and genetic fingerprint of each patient’s disease and could constitute a valid alternative to traditional biopsy for diagnosis,stratification, and treatment response [13].
The analysis of cell-free DNA (cfDNA) in plasma provides an opportunity for minimally invasive tumor profiling since a fraction of plasma cfDNA in cancer patients is tumor-derived (circulating tumor DNA [ctDNA]) [14]. When looking for single nucleotide variations(SNV) orsmall indel in tumors or ctDNA, the use of next-generation sequencing (NGS) panel assay with very high sequencing depth (5,000–12,000X) is needed, especially to confidently detect SNV belonging to low abundance clones [15]. Instead, for large structural variations, ctDNA fraction calculation, and CNA inference, performing whole-genome sequencing (WGS) is more suitable than targeted panels, due to the higher breadth of coverage (percentage of target bases that are sequenced). Since HCC has no known targetable mutations, the utility of NGS panels in clinical practice is limited. On the other hand, the majority of HCCs exhibit high chromosomal instability. This characteristic may have prognostic implications and has been studied using WGS in tumor tissue [16]. CNAs are significant subclasses of somatic mutations. They involve amplifications or deletions of large chromosomal regions, resulting in the overexpression of oncogenes or the loss of tumor suppressor genes, thereby promoting carcinogenesis [17]. Recently, WGS has been applied to study large structural variations and copy number alterations (CNA) in ctDNA. As an illustration, cfDNA WGS (with an approximate sequencing depth of five times the whole genome, or 5×) has shown promise in identifying clinically significant tumor genomic alterations [18,19]. However, conducting WGS at this depth entails substantial sequencing costs, which may render this approach less viable for routine clinical practice. Several studies have successfully employed low-pass whole-genome sequencing (LP-WGS) instead, which uses a lower depth coverage and cost. However, even with 1.5× depth coverage, the cost of this approach remains prohibitive for routine clinical practice [20]. To overcome this limitation, ultra-low-pass whole-genome sequencing (ULP-WGS) (≤0.5×) has emerged as a low-cost promising alternative to estimate ctDNA and tumor CNAs [20].
It has recently been reported that CNAs and ctDNA fraction correlate with tumor burden, progression-free survival (PFS), and overall survival (OS) in early HCC patients receiving radical treatments (surgery and radiofrequency ablation). Importantly, when WGS (5x depth) was used, similar patterns of CNAs were observed between plasma ctDNA and tumor tissue [18,19]. Similar results were reported in patients with advanced HCC who underwent transarterial chemoembolization (TACE) with an average deep coverage of 3x [21]. Still, the validity of LP-WGS or ULP-WGS, which could be affordable and reliable prognostic tools in patients with HCC, remains unexplored.
In the present study, we aimed to test the clinical impact of using ctDNA and CNA detection by ULP-WGS of plasma cfDNA as a blood-based biomarker to identify patients with HCC who carry a worse prognosis, including patients with advanced HCC undergoing systemic treatment.
MATERIALS AND METHODS
Study design
Blood samples were prospectively and retrospectively collected from patients with a diagnosis of HCC and patients with cirrhosis without HCC at the Liver Unit of Clinica Universidad de Navarra between 2017 and 2022. Samples of HCC patients were prospectivelly collected between November 2021 and November 2022. Samples from cirrhotic controls were obtained retrospectively between 2017 and 2022. Among these samples, two belonged to patients with HCC who underwent liver transplantation, and three were from patients with cirrhosis without HCC. Informed consent from the HCC patient group was obtained before treatment, which included surgery (liver transplantation and resection), locoregional therapies(transarterial radioembolization and ablative therapies), and systemic treatments (sorafenib and immunotherapy). Patients with cirrhosis without HCC provided informed consent during a regular follow-up visit. This study was approved by the Research Ethics Committee of the Universidad de Navarra. Samples and data from patients included in the study were provided by the Biobank of the University of Navarra and were processed following standard operating procedures approved by the Ethical and Scientific Committees. All patients underwent clinical management and follow-up in the HPB Oncology Area of Clínica Universidad de Navarra.
Blood sample processing and cfDNA extraction
Whole blood samples (10 mL) were collected in EDTA (BD Biosciences, San Jose, CA, USA) and centrifuged at room temperature (2,000×g for 10 minutes). Isolated plasma was centrifuged a second time at room temperature (2,500×g for 10 minutes) in LoBind Eppendorf tubes to remove residual cells. Purified plasma was frozen at –80˚C until cfDNA isolation. Purified plasma was thawed on ice, followed by a short centrifugation at 4˚C (11,000×g for 15 minutes). cfDNA was extracted using the QIAamp Circulating Nucleic Acid kit (Qiagen, Hilden, Germany). Extracted cfDNA concentration was measured using the Qubit dsDNA High-Sensitivity assay (Thermo-Fisher, Waltham, MA, USA). Extracted cfDNA was stored in LoBind Eppendorf tubes at –80°C until further analysis.
Library preparation and ultra-low pass whole-genome sequencing
Library construction of cfDNA was performed using the NEBNext Ultra II DNA Library Prep Kit (NEB) according to the manufacturer’s instructions. A total of 2.5 ng of cfDNA input was used for ULP-WGS. Sequencing libraries were pooled and sequenced with a NextSeq2000 (Illumina) using 100 bp paired-end runs with an average coverage of 0.3×.
Data analysis
Fastq files from the sequencing platform were quality filtered with TrimGalore, and sequences shorter than 50 bp were removed. Then, sequences are aligned with Bowtie2 using the hg19 database. Bam files were sorted and indexed with Samtools. Finally, duplicates were tagged using MarkDuplicates feature from Picard tools. Following the preprocessing, data were analyzed using ichorCNA package according to the workflow proposed by their developers. To identify large-scale CNAs and aneuploidies, we used the software package ichorCNA. IchorCNA uses a Hidden Markov Model to predict the segments of CNAs and to estimate the ctDNA fraction from ULP-WGS of cfDNA [14]. The workflow consists of three steps: (1) computing read coverage, (2) data normalization, and (3) CNA prediction and estimation of ctDNA fraction. The analysis proceeded with a series of steps aimed at enhancing the accuracy of the results. Initially, guanine-cytosine content and mappability bias correction, depth-based local copy number estimations, and the estimation of tumor fraction based on copy number were carried out using the ichorCNA tool. Local read depth was corrected, considering guanine-cytosine bias and identifying regions with low mappability. Additionally, artifacts were eliminated by comparing the data to ichorCNA’s integrated healthy control reference. The CNAs were predicted with specific parameters tailored to the sample type, including the recommended low tumor fraction parameters for cfDNA samples and the default parameters for tumor and germline samples. Subsequently, ichorCNA utilized these binned, bias-corrected copy number values to create a two-component model, distinguishing between tumor-derived and non-tumor-derived fragments. From this model, the fraction of reads originating from the tumor, referred to as the tumor fraction, was derived [14].
Statistical analysis
The patients were categorized into two groups (positive or negative) based on the presence or absence of detectable ctDNA. This binary predictor variable was tested for association with clinical and demographic features using the Fisher’s exact or chi-square tests and t-test as appropriate. Categorical variables were reported as frequencies and percentages, and continuous variables were reported as medians, ranges, or interquartile range. OS and PFS were estimated by the Kaplan–Meier method. The association between OS with ctDNA positivity was tested using the log-rank test. Cox proportional hazards models were used to assess the association of ctDNA with other prognostic factors in the group of patients receiving systemic treatment. All P-values were two-sided; a P-value <0.05 was considered statistically significant. All analyses were performed using SPSS 25.0 (IBM Co., Armonk, NY, USA).
RESULTS
Patient characteristics
Blood samples were obtained from 73 patients with HCC. Patients most frequently had non-viral etiology (65.8%) and were in Child-Pugh class A (76.8%) (Table 1). Half of the patients were in very early or early BCLC stage, and 42% received systemic therapy that was mostly sorafenib. The median follow-up period was 37.38 months (range 0.5–65.6 months), and 25 patients (34.2%) had died at the time of analysis. As control group, blood samples were also obtained from 41 patients with cirrhosis without HCC or another malignancy. Most patients had non-viral etiology (75.6%) and were in Child-Pugh class B (70.7%). None of these patients developed malignancy during a median follow-up period of 22.47 months(range 1 to 58.4 months) (Table 2).

Characteristics of HCC patients
Characteristics of cirrhotic patients
Circulating tumor DNA is detected by ULP-WGS in HCC patients but not in cirrhotic patients
The median cfDNA concentration was 33.7 ng/mL (range 6.05–495) in the HCC cohort and 31.95 ng/mL (range 17–345) in the cohort of patients with cirrhosis. ctDNA was detected in 22 of 73 patients with HCC (30.1%), and the median percent of ctDNA fraction was 27% (range 14–70%). Among patients receiving systemic treatment, ctDNA was detected in 18 of 31 patients (58.1%). Using ULP-WGS, we did not detect ctDNA in any of the patients in the cirrhotic cohort.
Detection of ctDNA is associated with clinical features and outcomes in HCC patients
We, therefore, analyzed the association between the presence of ctDNA with baseline clinical and laboratory features (Table 3). The patients who tested positive for ctDNA more frequently had advanced HCC, including BCLC B/C stage, macrovascular invasion, extrahepatic spread, larger tumor size, and high levels of AFP (≥20 ng/mL), compared to those who tested negative (P<0.05). There were no significant differences in age, sex, etiology (viral vs. non-viral), tumor number, and degree of involvement (bilobar vs. unilobar) between patients with and without detectable ctDNA.

Association between ctDNA detection and clinical features in patients with HCC
Patients were followed for a median of 37.4 months (range 0.53–65.64 months). Among patients receiving systemic treatment, the detection of ctDNA was associated with inferior OS (Fig. 1). Median OS was not reached in the ctDNA-negative group, while it was 13.9 months(95% confidence interval [CI] 6.46–21.46) in the ctDNA-positive group (P=0.01). PFS was also numerically lower for patients with detectable ctDNA, but the difference was not statistically significant (P=0.119). Median PFS was 8.7 months (95% CI 2.21–15.26) in the ctDNA-negative group and 4.2 months (95%CI 2.67–5.80) in the ctDNA-positive group.
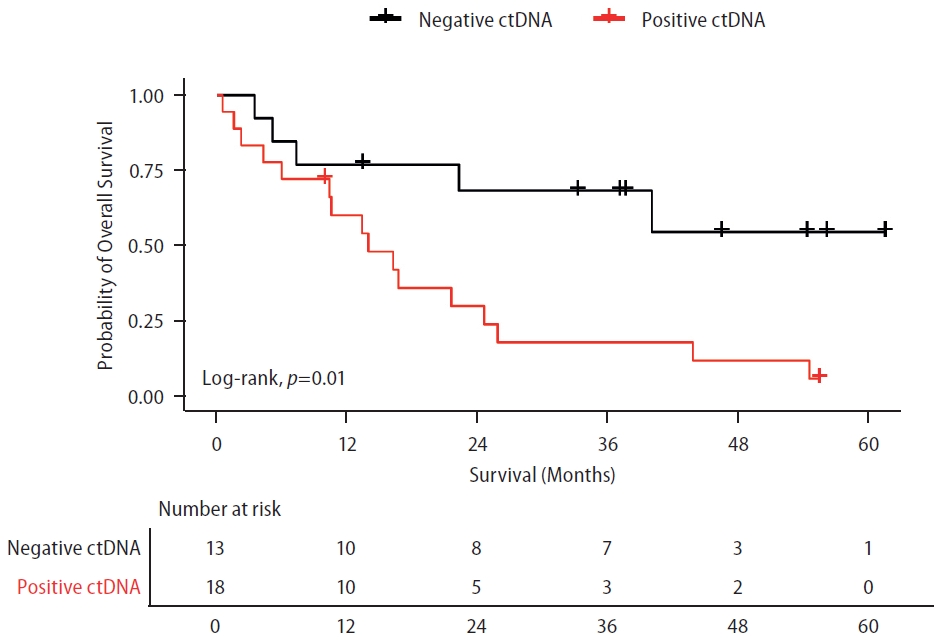
Overall survival according to ctDNA detection using ultra-low pass whole genome sequencing in patients with HCC under systemic treatment. Survival curve of patients undergoing systemic treatment according to the presence or absence of ctDNA. LogRank test was used for analyzing the median survival differences. Tick marks indicate censored data. ctDNA, circulating tumor DNA. ctDNA, circulating tumor DNA; HCC, hepatocellular carcinoma.
The four patients in the ctDNA-positive group, who did not receive systemic treatment due to being candidates to surgery or loco-regional therapy, were alive and free from relapse at the end of follow-up. Two of them were treated with hepatectomy (BCLC-A stage and BCLC-0 stage), and the other two were treated with radioembolization (BCLC-A stage and BCLC-B stage).
After adjusting for factors such as macrovascular invasion, extrahepatic spread, tumor size ≥5 cm, AFP ≥20 ng/mL, and type of treatment (sorafenib vs. immunotherapy), a multivariable Cox proportional hazard regression analysis revealed a significant association between the presence of ctDNA and OS in patients with HCC receiving systemic treatments. The hazard ratio (HR) was 7.69 (95% CI, 2.09–28.27). HRs for macrovascular invasion, extrahepatic spread, tumor size ≥5 cm, AFP ≥20 ng/mL, and treatment with sorafenib were 1.84 (95% CI, 0.67–5.02), 0.35 (95% CI, 0.09–1.24), 1.12 (95% CI, 0.33–3.83), 1.61 (95% CI, 0.38–6.77), and 4.92 (95% CI, 1.54–15.67), respectively. Treatment with immunotherapy was protective (HR 0.20; 95% CI 0.06–0.64) (Supplementary Table 1).
The percentage of ctDNA among the ctDNA-positive patients was not associated to OS, but the sample size was low (P=0.142), with a median survival of 13.37 months (95% CI, 9.45–17.28) for those with high ctDNA fraction compared to 21.61 months (95% CI, 7.41–35.82) for those with low ctDNA fraction.
In a subgroup analysis based on the BCLC stage, a non-significant trend towards worse OS in the presence of ctDNA was observed in the more advanced stages. No BCLC-0 patient and only two BCLC-A patients had died at the end of follow-up. Among 14 BCLC-B patients (six ctDNA-positive), the median overall survival was not reached in the ctDNA-negative group and was 13.9 months in the ctDNA-positive group (P=0.184). Among 22 BCLC-C patients (13 ctDNA-positive), the median overall survival was 40.1 months in the ctDNA-negative group and 16.3 months in the ctDNA-positive group (P=0.148).
Identification of genetic features of HCC using ULP-WGS
ULP-WGS data analysis showed CNAs at different chromosomal loci. According to the previously described structural genomic variations in HCC sequencing [22], certain chromosomal alteration patterns were commonly found in patients with HCC. The most frequent chromosomal arm gains were 1q (63.6%), 8q (59.1%), 7q (27.2%), and 5p (22.7%), and the most frequent chromosomal arm losses were 8p (54.5%), 4q (45.4%), 13q (45.4%), 16q (40.9%), and 5q (36.3%) (Fig. 2A). The frequency of CNA per patient was highly variable, and a minority of patients’ ctDNA had only one chromosomal arm affected. While 22.7% of patients had only one gain, another 22.7% had more than seven gains (Fig. 2B). In the case of losses, 9.1% of patients had one loss, while 50% had more than seven losses (Fig. 2C). Considering either gains and losses, only 18.2% of patients had only one gain or loss, while 59.1% had more than seven chromosomal arm gains or losses (Fig. 2D).
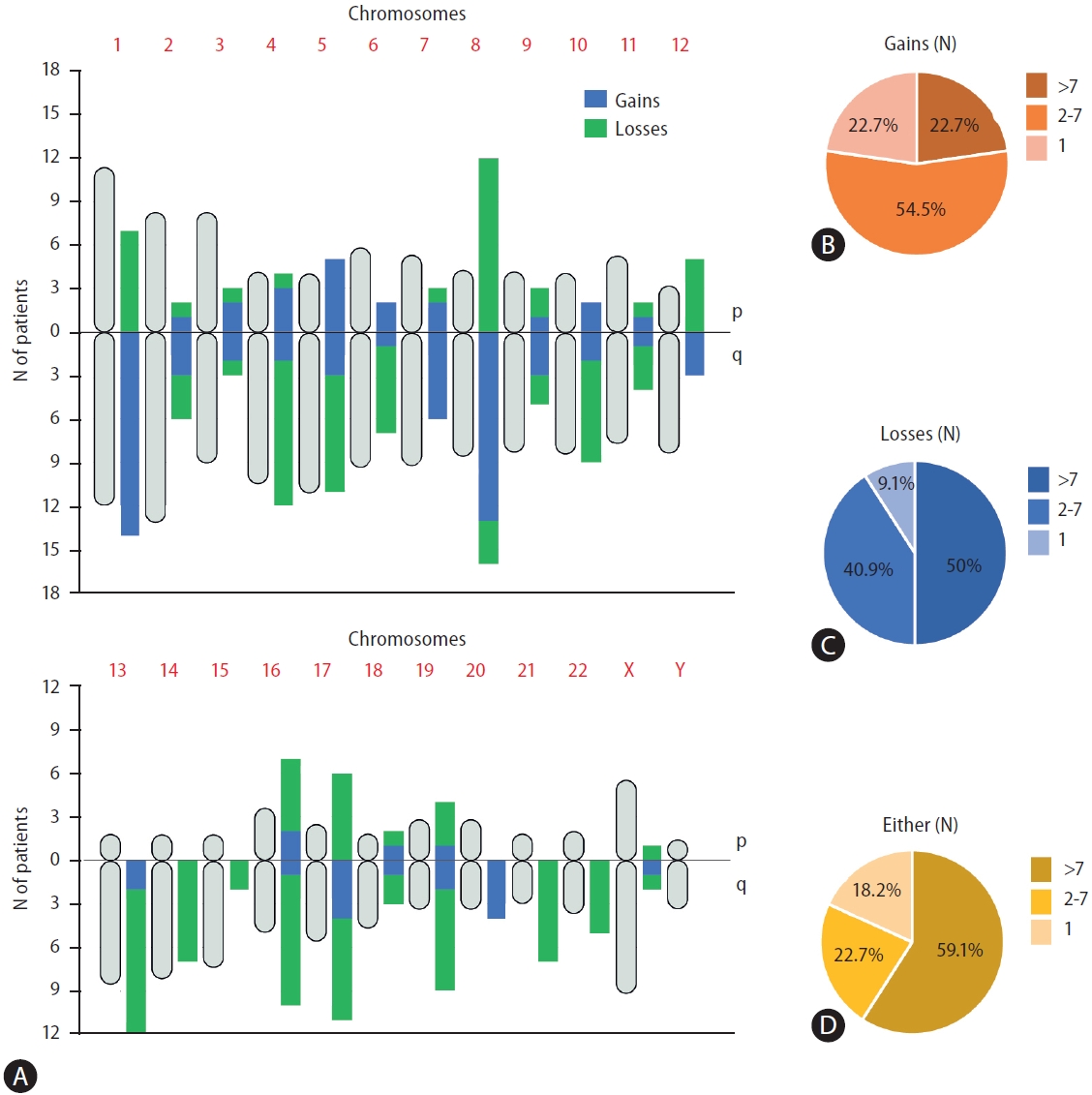
Distribution of large copy number alterations along the whole genome and the cohort. (A) Diagram showing the relative length of each chromosome arm in the human genome (gray rounded shaped vertical symbols) with, besides, the number of patients with either gains (blue) and losses (green) in each chromosome arm. (B) Percentage of patients with a specific number of concomitant CNAs gains in different arms. (C) Percentage of patients with a specific number of concomitant CNAs losses in different arms. (D) Percentage of patients with a specific number of concomitant CNAs losses in different arms. CNA, copy number alterations.
In ctDNA-positive patients, more than seven CNA (either gains or losses) was associated with inferior OS. Median OS was 54.6 months (95% CI 21.06–88.21) in the less than seven CNA group, and 10.5 months (95% CI 3.28–17.80) in the more than seven CNA group (P=0.006).
There was no difference in the OS of patients with gain of 1q, 8q, 7q, or 5p compared to those without a corresponding chromosomal-arm gain. Among the patients under systemic treatment with positive ctDNA, those with loss of 5q and 16q exhibited a significantly worse OS compared to those without a corresponding chromosomal-arm loss (both P<0.05). The median OS was 10.38 months (95% CI, 0–22.91) with 5q loss and 21.61 months (95% CI, 10.09–33.13) without the loss (Fig. 3A). The median OS was 5.97 months (95% CI, 0.98–10.97) with 16q loss and 21.61 months (95% CI, 7.41–35.82) without it (Fig. 3B).
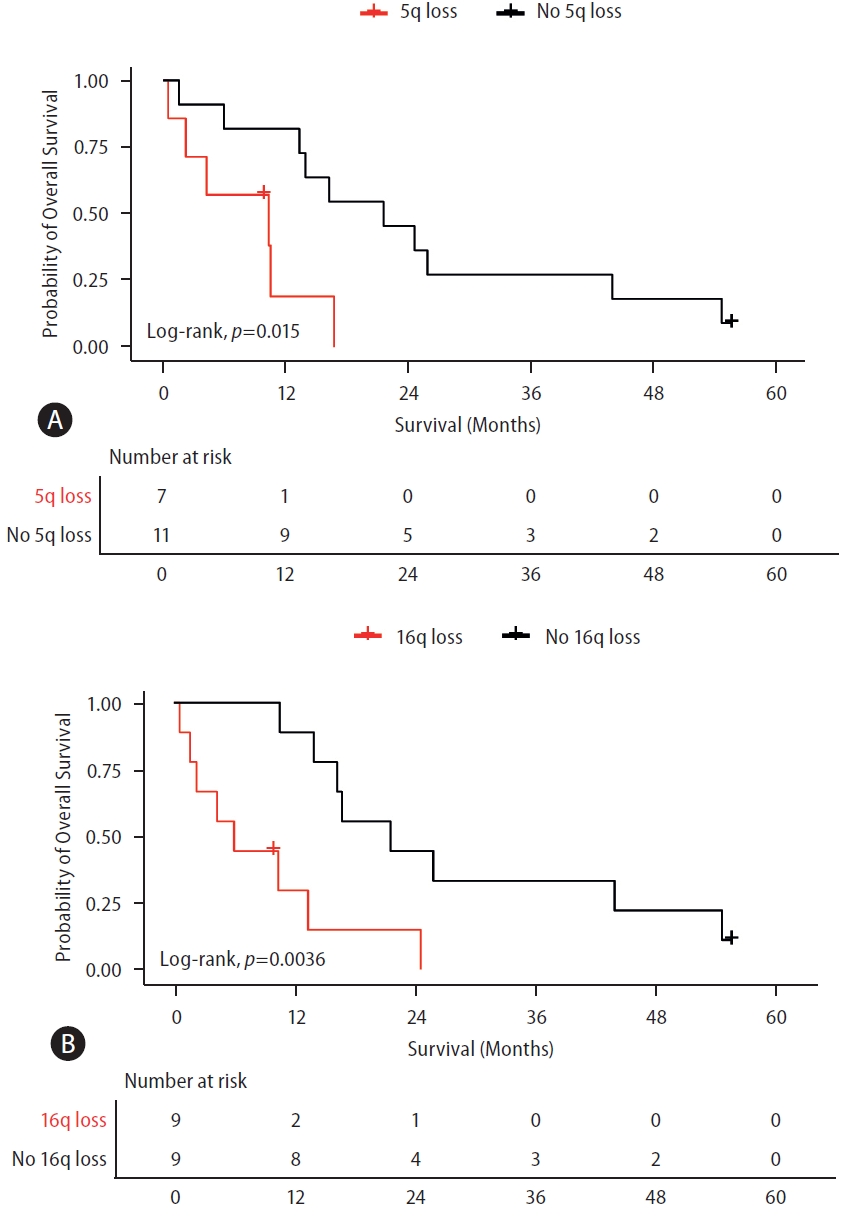
Overall survival of patients with advanced HCC according to prognostically-relevant CNAs. (A) Survival curve of patients with detectable ctDNA according to the presence or absence of 5q loss. (B) Survival curve of patients with detectable ctDNA according to the presence or absence of 16q loss. All patients were treated with systemic treatments. LogRank test was used for analyzing the median survival differences. Tick marks indicate censored data. 5q, long arm of chromosome 5; 16q, long arm of chromosome 16. ctDNA, circulating tumor DNA; HCC, hepatocellular carcinoma; CNA, copy number alterations.
A significant association was found between 5q loss and OS after adjusting for AFP ≥20 ng/mL, macrovascular invasion, tumor size ≥5 cm, and extrahepatic spread. HR for 5q loss was 8.92 (95% CI, 1.79–44.38). HRs for AFP ≥20 ng/mL, macrovascular invasion, tumor size ≥5 cm, and extrahepatic spread were 0.98 (95% CI, 0.17–5.70), 0.51 (95% CI, 0.14–1.80), 2.52 (95% CI, 0.55–11.40), and 0.30 (95% CI, 0.06–1.43), respectively (Supplementary Table 2).
Moreover, an independent association was observed between 16q loss and the OS, after adjusting for variables such as AFP ≥20 ng/ml, macrovascular invasion, tumor size ≥5 cm, and extrahepatic spread. HR for 16q loss was 5.29 (95% CI, 1.24–22.50). HRs for AFP ≥20 ng/mL, macrovascular invasion, tumor size ≥5 cm, and extrahepatic spread were 0.39 (95% CI, 0.06–2.47), 1.31 (95% CI, 0.32–5.33), 2.30 (95% CI, 0.49–10.84), and 0.43 (95% CI, 0.07–2.49), respectively (Supplementary Table 3).
DISCUSSION
Based on our results, detectable ctDNA serves as a minimally invasive biomarker indicating a worse prognosis in patients with HCC undergoing systemic therapy, independent of clinicopathologic characteristics and type of systemic treatment. Patients with detectable ctDNA were more likely to exhibit unfavorable biological tumor features, including AFP ≥20 ng/mL, macrovascular invasion, tumor size ≥5 cm, type of systemic treatment, and extrahepatic spread.
Our results are supported by recent studies that demonstrated the association of ctDNA detected by ULP-WGS with poor OS in various cancer types, including metastatic squamous non–small cell lung cancer [23], Ewing sarcoma, and osteosarcoma [24], metastatic castration-resistant prostate cancer [25], cervix cancer [26], and metastatic triple-negative breast cancer [27]. Our study is the first to show the same phenomenon in advanced HCC patients. The identification of ctDNA is a biomarker for tumor aggressiveness and may allow more accurate risk stratification, treatment planning, and surveillance.
ULP-WGS presents several advantages. Among them, easy processing, low cost, and rapid readout standout as the most relevant advantages for routine clinical practice. If confirmed in larger series, ULP-WGS could help in prognostic assessment. Tumor staging with clinical and imaging features allow allocating patients in groups with different prognosis [3]. AFP and AFP-L3% can identify a group of patients with worse prognosis across stages [28]. However, accurate individual prognostication is still an unmet need in HCC. Contrary to AFP measurement, ULP-WGS is not focused on a specific type of genetic alteration. It can identify and group various genetic alterations, contributing to a reduction in result variability. Notably, detecting ctDNA and CNAs with this approach possesses a distinctive capacity to encapsulate comprehensive somatic information about HCC. This unique attribute may overcome those challenges related to tumor heterogeneity. Tissue biomarkers, such as Heat Shock Protein 70, can offer diagnostic and prognostic utility [29,30]. However, accessing this information requires biopsies or surgical specimens, which come with inherent risks. ULP-WGS could also potentially help in monitoring the response to treatment and provide a dynamic picture of the disease course.
Limitations of ULP-WGS include its lower sensitivity and the need of a relatively high tumor burden for effective detection of ctDNA and CNAs, as shown by the poor performance in BCLC 0/A patients. In such earlier stages or for the detection of minimal residual disease, more sensitive methods would be needed, such as the detection of tumor-derived SNV using Droplet Digital PCR or deep-targeted sequencing [20]. Studies in prostate cancer have also shown that ctDNA was not detected in patients with local vs. metastatic disease [25,31]. The potential mechanisms include reduced necrosis and vascularization of localized small tumors with diminished proliferative rate [31,32]. Understanding the strengths and limitations of ULP-WGS underscores the importance of tailoring the approach to the specific clinical context and disease stage for optimal utility.
The utility of detecting ctDNA using cfDNA WGS in patients with HCC has been reported in a cohort of 117 subjects with early tumors receiving surgery or radiofrequency ablation [19]. In this cohort, the sequencing depth coverage of the WGS was relatively high (5×). Higher levels of ctDNA were associated with poor recurrence-free and OS. The most frequent CNAs included gains in 20p, 8q, 1q, and 20q, as well as losses in 17p, 4q, 19p, and 16q [19]. In another cohort of 34 HCC patients undergoing surgery, ctDNA detected by WGS (with a deep coverage of 5×) had prognostic value [18]. Similar findings were observed in a longitudinal cohort of 64 subjects with advanced HCC receiving TACE. In this cohort, the average depth of sequencing coverage was 3x. Notably, they found that the changes in ctDNA during TACE treatment correlated with tumor burden and had predictive value for treatment response and prognosis. The most common CNAs were gains in the regions of chromosomes 1q, 6p, 8q, 20q, and 20q along with losses in chromosomes 4q, 13q, 8p, 16q, and 17p [21].
In the present study, we discovered various genomic features in ctDNA that were prognostically relevant, including the detection of CNAs at different chromosomal loci in HCC. Among patients with detectable ctDNA undergoing systemic therapy, the loss or deletion of 5q and 16q emerged as independent biomarkers predicting worse survival, regardless of other clinicopathologic features usually associated with bad prognosis,such as high AFP, macrovascular invasion, or extrahepatic spread. Furthermore, most patients exhibited more than seven concurrent CNAs, indicating a high chromosomal instability and molecular heterogeneity, which ultimately lead to disease progression.
In our study, the most frequent losses were 8p, 4p, 13q, 16q, and 5q, all reported as frequent in HCC, in large cohort studies involving the WGS of tumor samples [33]. The loss of 5q and 16q chromosome arms was observed in a group of patients with poor survival. The loss at 5q was reported in HCC [16,34,35]. Notably, the genomic loci of 5q13.2 encompass cancer related genes, including GTF2H2, NAIP, and OCLN [34]. In a previous study involving 29 HCC patients, the loss of 5q was observed in the tissue of nine patients (31%). Multivariate analysis revealed that allelic loss on chromosome 5q34 band served as an independent prognostic factor for poor survival [16]. On the order hand, the long arm of chromosome 16 carries the epithelial cadherin (E-cadherin) gene, a finding supported by previous studies in HCC [17,36,37]. E-cadherin is a cell adhesion protein implicated as an invasion and metastasis suppressor. A meta-analysis involving 2,439 patients demonstrated that reduced expression of E-cadherin correlated with a poor prognosis in HCC. It is also associated with metastasis, vascular invasion, advanced differentiation grade, and advanced disease stage [36].
The three most frequent losses at chromosomes 8p, 4p, and 13q were not associated with survival. Chromosome 8p harbors a cluster of six genes, including DLC1, CCDC25, ELP3, PROSC, SH2D4A, and SORBS3, all of which are tumor suppressor genes [33]. Also, the inhibitor of growth family member 2 (ING2) is a tumor suppressor gene located on chromosome 4q [17,22]. Frequent allelic losses at chromosome 13q have been observed in HCC [33]. The retinoblastoma gene (RB1), located in this chromosome, is believed to play a role in HCC [38].
In our cohort, the most frequent gains were 1q, 8q, 7q, and 5p. These amplifications encompass well-known driver oncogenes, including MCL1 (1q21.3), MET (7q31.2), MYC (8q24.21), and TERT (5p15.33) [22].
CNAs are important subclasses of somatic mutations, with aberrant chromosomal regions of amplifications or deletions commonly associated with overexpressed oncogenes or the loss of tumor suppressor genes. CNAs are a hallmark of human cancer and are believed to contribute to carcinogenesis, tumor progression, and the development of therapy resistance [39,40]. In a previous study involving patients with metastatic prostate and breast cancer, tumor-derived CNAs were detected in ctDNA using ULP-WGS, and these were found to be concordant with those observed in the corresponding tumor tissue [14].
Our study has some limitations. First, the technique used to assess ctDNA and CNAs exhibited low sensitivity, particularly in patients at early stages. Moreover, the retrospective nature of the study raises concerns about potential bias. Another limitation include the heterogeneity of treatments: the majority of patients received sorafenib and the patients treated with immunotherapy received different regimes. Consequently, a validation cohort with a larger sample size and consecutive blood samples, ideally involving patients undergoing first-line systemic treatments such as combination therapies of Atezolizumab-Bevacizumab or Durvalumab-Tremelimumab, would be highly valuable.
In summary, our study explores the utility of using ULP-WGS of cfDNA to detect the presence of ctDNA and CNAs in HCC. Our findings demonstrate that the detection of ctDNA and CNAs can provide clinically relevant information regarding HCC prognosis. The assessment of ctDNA and CNAs has the potential to serve as a prognostic biomarker in advanced HCC, to help provide better information about unique genomic features, tumor progression, drug resistance, and novel therapeutic targets.
Notes
Authors’ contribution
Conceptualization: J.A., B.S., and M.S; Writing the original draft: M.S., B.S., and J.A.; DNA extraction: M.A., C.B, and I.B, Genomic analysis: F.M., J.Z., and A.dV.; Bioinformatics: M.P.; Review & editing: M.A., C.B., D.D., and M.I.
Conflicts of Interest
BS reports consultancy fees from Adaptimmune, Astra Zeneca, Bayer, BMS, Boston Scientific, Eisai, Eli Lilly, Incyte, Ipsen, Novartis, MSD, Roche, Sanofi, Sirtex Medical, Terumo; speaker fees from Astra Zeneca, Bayer, BMS, Eisai, Eli Lilly, Incyte, Ipsen, Novartis, Roche, Sirtex Medical, Terumo; research grants (to the Institution) from BMS and Sirtex Medical. J.A. is member of a scientific steering committee of Roche Spain and has received fees for talks by Pfizer and Roche. Rest of the authors have nothing to declare.
Acknowledgements
We particularly acknowledge the contributions of the patients for their participation and the Biobank of the University of Navarra for its collaboration, as well as Amaya Redín from the Liver Unit of Clínica Universidad de Navarra.
Instituto de Salud Carlos III, Accion Estrategica en Salud (AES2020-2023): PI20/1663 grant to JA.
Fundacion Echebano grant 2022-2023 grant to JA.
Abbreviations
HCC
hepatocellular carcinoma
BCLC
Barcelona Clinic Liver Cancer
AFP
alpha-fetoprotein
cfDNA
cell-free DNA
ctDNA
circulating tumor DNA
WGS
whole-genome sequencing
LP-WGS
low-pass whole-genome sequencing
ULP-WGS
ultra-low-pass whole-genome sequencing
CNAs
copy number alterations
OS
overall survival
PFS
progression-free survival
IQR
interquartile range
HR
hazard ratio
CI
confidence interval
SUPPLEMENTAL MATERIAL
Supplementary material is available at Clinical and Molecular Hepatology website (http://www.e-cmh.org).
Multivariable Cox proportional hazards model for patients receiving systemic treatment
Multivariable Cox proportional hazards model for patients under systemic treatment with positive ctDNA with loss of 5q
Multivariable Cox proportional hazards model for patients under systemic treatment with positive ctDNA with loss of 16q
References
Article information Continued
Notes
Study Highlights
• The detection of ctDNA using ultra-low-pass whole-genome sequencing carried worse prognosis in HCC patients under systemic treatment. Furthermore, the loss of the long arms of chromosomes 5 and 16 was associated with worse survival among ctDNA-positive patients receiving systemic treatment. Ultra-low-pass whole-genome sequencing may provide a relevant affordable tool to improve the prediction of prognosis in HCC, which is important for clinical research and practice.