New treatments for chronic hepatitis C
Article information
Abstract
Treatments for chronic hepatitis C has evolved significantly in the past 15 years. The standard of care (SOC) is peginterferon alfa-2a/-2b with ribavirin for 48 weeks or 24 weeks in patients infected with HCV genotype 1 or 2/3, respectively. The treatment duration can be individualized based on the baseline viral load and the speed of the virologic response during treatment. However, current therapies are associated with side effects, complications, and poor patient tolerability. Therefore, there is an urgent need to identify better strategies for treating this disease. An improved sustained virologic response (SVR) can be achieved with new HCV-specific inhibitors against NS3/4A and NS5B polymerases. Recent trials have found SVR rates in patients with HCV genotype 1 infection of 61~68% and 67~75% for combining the SOC with the protease inhibitors telaprevir and boceprevir, respectively. Several new HCV-specific inhibitors such as protease inhibitors and nucleoside and non-nucleoside polymerase inhibitors as well as non-HCV-specific compounds with anti-HCV activity are currently in clinical evaluation. In this review we discuss these new treatments for chronic hepatitis C.
INTRODUCTION
Since the identification and molecular cloning of hepatitis C virus (HCV) in the late 1980s, it has been estimated that more than 170 million people are infected with the virus. In approximately 80% of infections the virus is able to elude the body's immune response and succeeds in establishing a chronic infection.1 The number of individuals infected with HCV continues to increase and persistently infected persons are at risk of developing cirrhosis and hepatocellular carcinoma. The current standard of care for the treatment of HCV infection is a combination of pegylated interferon and ribavirin (Peg-IFN/RBV). Because of the adverse effects associated with both interferon (IFN) and ribavirin and because Peg-IFN/RBV provides only about a 45~50% sustained virological response (SVR, undetectable HCV RNA for greater than 24 weeks after cessation of therapy) in genotype 1-infected individuals, there is a need for more potent anti-HCV compounds with fewer adverse effects. We will discuss new therapies for chronic hepatitis C.
Key features of hepatitis C virus
In recent years there have been significant advances in our understanding of the replication of HCV and the role of viral non-structural proteins.2-4 HCV has a single-stranded positive sense, 9.6 kb RNA genome that is flanked at each terminus by a 5' and 3' non-translated region (NTR) and contains one long open reading frame that encodes a precursor polyprotein of about 3,000 amino acids (Fig. 1). Translation of the polyprotein is directed by the internal ribosome entry site located within the 5'-NTR. The polyprotein is subsequently processed into both structural (core, envelope 1, envelope 2) and non-structural (p7, NS2, NS3, NS4A, NS4B,NS5A, NS5B) proteins by cellular and viral proteases. Core protein is a highly basic protein that forms the nucleocapsid. The envelope proteins E1 and E2 are highly glycosylated transmembrane proteins that associate non-covalently to form the viral envelope. Protein p7 is a highly hydrophobic polypeptide that forms hexamers and has been reported to have ion channel activity. NS2 is a cis-acting autoprotease that is essential for viral replication. NS2 catalyzes the cleavage of the polyprotein precursor at the NS2/NS3 junction, and also plays an essential role in virus assembly. NS3 is a bifunctional protein with serine protease activity in the amino terminal one-third, which is responsible for cleavage at the NS3/NS4A, NS4A/NS4B, NS4B/NS5A and NS5A/NS5B sites, and NTPase/helicase activities in the C-terminal two-thirds of the protein. The NS4A polypeptide functions as a co-factor for NS3 protease activity. NS4B was demonstrated to induce specific cellular membrane changes, creating a membranous web that serves as a scaffold for the formation of the viral replication complex. NS5A is a phosphoprotein with multiple functions and is essential for viral replication and assembly. A potential role in modulating the IFN response has also been suggested for NS5A.NS5B catalyzes the synthesis of both minus-strand and plus-strand RNA.
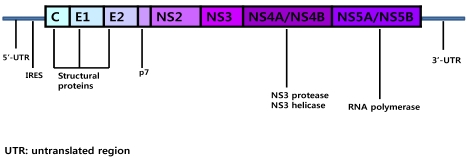
Schematic representation of the hepatitis C virus genome.
Hepatitis C and innate immunity
Increasing evidence suggests that HCV can interfere with innate immune activation at multiple levels. First, HCV, through its viral proteins, can undermine viral recognition by cleaving pivotal adaptor proteins in toll-like receptor (TLR3) and retionic acid-inducible gene-I (RIG-I) or melanoma differentiation-associated gene 5 (MDA5) signaling. Second, HCV directly or indirectly modulates key antigen-presenting functions of various dendritic cells (DC), contributing to impaired virus-specific T-cell activation. Third, IFN α production by plasmacytoid dendritic cells (pDCs), the main cell type producing IFNα, is drastically reduced in chronic HCV infection. Fourth, chronic HCV infection results in activation of proinflammatory pathways and mediators in inflammatory cells that contribute not only to aberrant innate-adaptive immune interactions but to activation of liver fibrosis and a microenvironment that may support cancer formation. Therapeutic strategies to counteract innate immune alterations in chronic HCV provide a promising target and require further investigation.5
Future therapies for HCV
Advances have been made in the development of new IFNs, specifically targeted therapy against hepatitis C (STAT-C) and host cell targets inhibiting HCV replication. This review focuses on recent clinical trials in that field.
1. New interferons
Currently, pegylated interferon α is the mainstay of HCV treatment regimens. However, several IFNs are under development and may offer improved responses, more convenient dosing regimens and/or improved tolerability. Albinterferon is a genetic fusion polypeptide of albumin and interferon alfa-2b with a longer half life than pegylated interferons. The phase 2 study comparing different doses of albinterferon alfa-2b and ribavirin with peginterferon alfa-2a and ribavirin indicated similar sustained virologic response rates with a better tolerability of albinterferon alfa-2b based treatment. Based on the encouraging findings from the phase 2 study, the efficacy and safety of albinterferon alfa-2b administered every two weeks in combination with ribavirin for 48 weeks and 24 weeks in patients infected with HCV genotype 1 and 2/3, respectively, was investigated in two phase 3, randomized, active controlled, multi-center studies.6,7 Both studies (ACHIEVE-1 and ACHIEVE-2) were designed to demonstrate non-inferiority of the albinterferon alfa-2b regimes compared with peginterferon alfa-2a. Both studies achieved the primary objective. In the intention-to-treat population, the sustained virologic response rates in the peginterferon alfa-2a, albinterferon alfa-2b 900 µg and albinterferon alfa-2b 1,200 µg groups were 51.0%, 48.2%, and 47.3% in patients infected with HCV genotype 1 and 84.8%, 79.8%, and 80.0% in patients infected with HCV genotype 2,3 respectively. The overall incidence of adverse events, serious or severe adverse events in the phase 3 studies was similar between the two treatments, indicating that albinterferon alfa-2b is not better tolerated than peginterferon alfa-2a. Locteron is a controlled-release interferon alfa-2b which is injected every 2 weeks. In a short term study controlled release interferon alfa-2b showed less flu like symptoms than peginterferon alfa-2b injected every week indicating that the controlled-release formulation may have a better tolerability. Larger trials powered to examine adverse event profiles and antiviral activity are being initiated.8 Peginterferon-λ is a pegylated type III interferon that binds to a unique receptor with more limited distribution than the type I IFN receptor. In a phase 1 healthy volunteer study, peginterferon-λ was pharmacologically active without flu-like symptoms or hematologic side-effects. In a phase 1b study the mean decline of HCV-RNA in patients with relapsed HCV genotype 1 infection was 1.9-3.6 log10 IU/mL after 4 weeks of retreatment with peginterferon-λ. Peginterferon-λ is currently being investigated in combination with ribavirin.9 Overall, the new IFNs may improve convenience and tolerability of IFN-based therapy. However, the current results on viral efficacy indicate that response rates will not be dramatically improved by the new IFNs.
2. Specifically targeted therapy against hepatitis C virus
The most attractive targets for future anti-HCV agents are those that specifically target the viral replication cycle (Fig. 2): internal ribosome entry site (IRES) inhibitors, viral protease inhibitors and transcription (polymerase) inhibitors and virus assembly inhibitors (Fig. 3). IRES inhibitors are RNA structures that lead to inhibition of HCV polyprotein translation. It is known that translation of HCV RNA is initiated by internal entry of ribosomes into the 50 non-coding region or IRES in vitro.10 IRES inhibitors include anti-sense oligonucleotides (e.g., ISIS 14803; Isis Pharmaceuticals, Carlsbad, CA), small interfering RNAs (siRNA) and ribozymes. To date, the clinical experience with oligonucleotides has been disappointing, due to evidence of toxicity11 and poor antiviral activity. A phase I study with ISIS 14803 demonstrated HCV reductions in only 3/28 patients treated, and further development of this compound is unlikely.11 New attempts are underway to find more efficient methods to inhibit the IRES. The most promising recent advances have come in molecules that target the NS3/4A serine protease and the NS5B RNA-dependent RNA polymerase. The structural solution of the NS3/4A protease and the HCV NS5B polymerase and the development of a (sub)genomic replicon system have enabled the development and testing of HCV specific compounds. Further attractive targets within the HCV genome for antiviral therapy are the envelope proteins which are involved in HCV entry and NS5A which is involved in replication and assembly and quite possibly in mediating IFN alfa resistance. The clinical development of NS3/4A protease inhibitors is currently most advanced.

Life cycle of the hepatitis C virus.
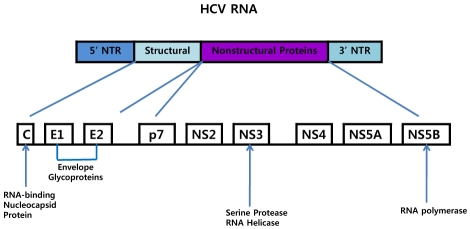
Different target regions in the hepatitis C virus genome.
1) Protease Inhibitors
NS3-4A Protease
The HCV RNA genome is translated as a single polyprotein precursor that is enzymatically processed by host and viral proteases.12 The N-terminal portion of the polypeptide is processed by host cell proteases to generate 3 structural proteins (core, the E1 and E2 envelope glycoproteins) and p7 (an integral membrane protein whose function has not yet been definitively determined). The remainder of the polypeptide contains 6 non-structural (NS) proteins required for viral replication and maturation, including NS3, a multifunctional enzyme with an N-terminal serine protease domain and a C-terminal RNA helicase/NTPase domain. With the addition of the NS4A cofactor essential for complete peptidase activity, the heterodimeric NS3-4A protease has substrate specificity distinct from that of host cell or other viral proteases. Early in vitro and primate studies demonstrated the essential role of the NS3-4A protease and highlighted the therapeutic potential of an HCV protease inhibitor.13,14 Chimpanzees inoculated with HCV clones with abrogated NS3-4A activity failed to generate productive HCV infection, suggesting that this protease is integral to viral replication and polypeptide maturation.14 Furthermore, in vitro data have demonstrated that the NS3-4A protease may participate in host immune evasion by targeting for degradation several key cellular signaling molecules associated with endogenous IFN production and responsiveness.15,16 The NS3-4A protease, therefore, may represent a dual therapeutic target by inhibiting viral replication and potentially restoring the innate response to chronic HCV infection.
Several protease inhibitors were investigated in clinical trials. Monotherapy with the protease inhibitors ciluprevir, telaprevir and boceprevir was shown to be effective in lowering the viral load. The development of ciluprevir was stopped due to cardiotoxicity in animal studies. Clinical evaluation of telaprevir and boceprevir is most advanced. Both protease inhibitors showed a rapid selection of drug resistant HCV strains within 2 weeks of therapy, indicating that protease inhibitor monotherapy will not suffice for treatment of patients with chronic hepatitis C. Because peginterferon alfa/ribavirin has a completely different mode of action and resistance profile than protease inhibitors and are active against protease-resistant variants, the current protease inhibitors are being investigated in combination with peginterferon with and without ribavirin.
(1) Telaprevir
The peptidomimetic inhibitor of the NS3/4A serine protease telaprevir showed a 3 log10 IU/mL decline of HCV RNA during the first 2 days of monotherapy in patients infected with HCV genotype 1 and previous non-response to IFN based antiviral treatment. However, during 14 days of monotherapy, a continuous decline of HCV RNA was noted in only 7 of 28 patients (25%). Using a highly sensitive sequencing method several mutations associated with resistance to telaprevir were identified. Mutations associated with resistance occurred in the NS3 catalytic domain either as single mutation (V36A/M, T54A, R155K/T, A156S/T/V) or as double mutation (at positions 36+155 or 36+156). Low level resistance mutations (V36A/M, T54A, R155K/T, and A156S) and high level resistance mutations (A156V/T, 36+155, 36+156) can be distinguished. Combination therapy of telaprevir with peginterferon alfa-2a and ribavirin was effective in preventing the rapid occurrence of resistance. The combination therapy of peginterferon alfa-2a/ribavirin/telaprevir was investigated in the PROVE1 and 2 studies.17,18 Both studies are complete and telaprevir is one of the first STAT-C compound for which sustained virologic response rates have been reported for combination therapy with peginterferon alfa-2a and ribavirin. In both trials triple therapy was given for 12 weeks. The sustained virologic response rates in PROVE1 and PROVE2 were 61% and 68% in patients treated with peginterferon alfa-2a/ribavirin/telaprevir for 12 weeks followed by peginterferon/ribavirin for 36 or 12 weeks, respectively. The sustained virologic response rates in these telaprevir arms were significantly higher compared with the sustained virologic response rates in the standard of care control arms (41% and 46% in PROVE1 and PROVE2 respectively). Overall, the PROVE-studies confirm that protease inhibitors are able to increase sustained virologic response rates in patients with HCV genotype 1 infection. Furthermore, the PROVE2 study indicates that by addition of telaprevir to SOC (the standard of care) higher sustained virologic response rates can be achieved with shorter treatment duration. The high antiviral efficacy of telaprevir in combination with IFN alfa raises the question whether ribavirin is still necessary in the era of protease inhibitors and if double combination with peginterferon and a protease inhibitor is sufficient for a sustained virologic response. In PROVE2 the sustained virologic response rate in patients treated with telaprevir/peginterferon alfa-2a without ribavirin for 12 weeks was lower than in patients treated with telaprevir/peginterferon alfa-2a plus ribavirin for 12 weeks (36% vs. 60%). The lower rate of sustained virologic response in the group without ribavirin was due to a higher relapse rate compared to the groups with ribavirin (48% vs. 14-29%). The results of the PROVE2-trial provide evidence that ribavirin has additive antiviral activity to telaprevir and peginterferon alfa-2a and that triple therapy is required for optimal sustained virologic response rates. Telaprevir in combination with peginterferon alfa-2a and ribavirin was also investigated in patients with prior non-response to standard of care. The PROVE3 trial was a randomized, placebo-controlled phase 2 study assessing safety and efficacy of telaprevir plus peginterferon alfa-2a±ribavirin in HCV genotype 1 patients who previously failed peginterferon/ribavirin treatment.19 The overall sustained virologic response rates were significantly higher in the telaprevir arms (peginterferon alfa-2a/ribavirin/telaprevir for 12 or 24 weeks followed by peginterferon alfa-2a/ribavirin for 12 and 24 weeks, respectively) compared with the control arm. Superior sustained virologic response rates were observed in the triple therapy arms compared with the SOC control arm or the peginterferon/telaprevir arm without ribavirin (38~39% vs 9~10%). Overall, the study provides evidence that protease inhibitors in combination with standard of care will be a viable treatment option for patients who failed previous antiviral therapy.
(2) Boceprevir
Boceprevir, another NS3/4A serine protease inhibitor, binds reversibly to the NS3 protease active site and has potent activity in the replicon system alone20 and in combination with IFN alfa-2b.20 In a phase 1 open-label combination study, boceprevir was evaluated in combination with pegylated interferon-alfa-2b versus either agent alone in a crossover design in adult patients who have HCV genotype 1 and were previous non-responders to pegylated interferon-alfa-2b-based therapy.21 Patients were randomized to receive in random sequence (1) boceprevir (200 mg or 400 mg every 8 hours) as monotherapy for 7 days, (2) pegylated interferon-alfa-2b (1.5 mg/kg/wk) as monotherapy for 14 days, and (3) boceprevir plus pegylated interferon-alfa-2b combination therapy for 14 days in a three-period crossover design with a 3-week washout between treatments. Mean maximum log10 changes in HCV RNA were -2.45±0.22 and -2.88±0.22 for pegylated interferon-alfa-2b plus boceprevir at a rate of 200 mg or 400 mg, respectively, compared with -1.08±0.22 and -1.61±0.21 for boceprevir at a rate of 200 mg and 400 mg, respectively, and -1.08±0.22 and -1.26±0.20 for pegylated interferon-alfa-2b alone in the groups with boceprevir administered at 200 mg and 400 mg, respectively.21 As for telaprevir several mutations associated with resistance were identified during boceprevir monotherapy (V36, T54, R155, A156, V170, V55A).22 Boceprevir shows an overlapping resistance profile with telaprevir, indicating that a combination therapy of both protease inhibitors will not be promising. Recently, the final results of the HCV SPRINT-1 study assessing safety and efficacy of boceprevir in combination with peginterferon alfa-2b (1.5 µg/kg/week) and ribavirin in treatment naïve patients with chronic hepatitis C genotype 1 infection were presented.23 The triple combination arms with a total treatment duration of 48 weeks with or without a 4 weeks peginterferon-alfa2b/ribavirin lead-in were associated with significantly higher sustained virologic response rates than the low dose ribavirin arm and the standard of care control arm (75% and 67% vs. 36% and 38%, respectively). The results from the PROVE and the SPRINT trials confirm the concept that specific protease inhibitors are able to improve the cure rates of patients with chronic hepatitis C. Furthermore, both trials indicate that ribavirin is still necessary for achieving a sustained virologic response.
(3) New protease inhibitors
ITNM-191, SCH 900518, TMC435, BI201335 and MK-7009 are novel NS3/4A protease inhibitors currently in clinical trials. Sustained virologic response rates are not available so far. ITMN-191 is a potent HCV NS3/4A protease inhibitor that achieves high liver concentrations following oral administration.24 ITNM-191 in combination with peginterferon alfa-2a/ribavirin showed a stronger decline of HCV RNA compared with peginterferon alfa-2a/ribavirin standard of care after two weeks of treatment (4.7-5.7 log10 IU/mL vs 2.0 log10 IU/mL). After 2 weeks, 13-57% of patients in the triple therapy arm while no patient in the standard of care arm showed undetectable HCV RNA. SCH 900518 with and without ritonavir boosting showed robust reductions in HCV RNA levels in both treatment-experienced and naïve HCV genotype 1-infected patients (4.01 log10 IU/mL and 4.5 log10 IU/mL vs 0.09-0.19 log10 IU/mL after 8 days in patients treated with SCH 900518 400 mg twice/day plus peginterferon alfa-2a/ribavirin plus ritonavir 100 mg/d and SCH 900518 800 mg thrice/day plus peginterferon alfa-2a/ribavirin, respectively, vs. patients receiving peginterferon alfa-2a/ribavirin alone).25 TMC435 administered for 4 weeks in combination with peginterferon-alfa-2a/ribavirin was well tolerated and demonstrated potent antiviral activity in HCV genotype 1 infected treatment-experienced patients (4.3-5.3 log10 IU/mL in the TMC435 arms vs. 1.5 log10 IU/mL in the control arms).26 BI 201335 was investigated as monotherapy for 14 days and in combination with peginterferon alfa-2a/ribavirin for 28 days in experienced patients and showed a median HCV RNA decline of 3-4.2 log10 IU/mL in monotherapy and 4.8-5.3 log10 IU/ml in combination therapy.27-29 MK-7009 is a non-covalent competitive inhibitor of HCV NS3/4A protease. In treatment naïve patients MK-7009 was administered for 28 days in combination with pegylated interferon-alfa/ribavirin. The rapid virologic rate was higher in patients treated with triple therapy than in patients treated with standard of care (68.8~82.4% vs. 5.6%).30 All new compounds were relatively safe and well tolerated in monotherapy as well in combination with standard of care and will be further developed for HCV treatment.
2) Polymerase inhibitors
Nucleoside analog versus Non-nucleoside polymerase inhibitors
Viral polymerase inhibitors are currently the largest class of antiviral drugs for the treatment of hepatitis B, HIV, and herpes viruses, and were some of the first drugs developed for the treatment of HCV. A large proportion of these drugs are nucleoside analogs, synthetic compounds structurally similar to nucleosides, the building blocks of RNA and DNA. The NS5B polymerase inhibitors for treatment of chronic hepatitis C are consist of 2 classes: (1) nucleoside or nucleotide inhibitors (active site inhibitors) and (2) non-nucleotide inhibitors (allosteric inhibitors). Nucleoside inhibitors are analogs of natural substrates of the polymerase that are incorporated into the growing RNA chain leading to chain termination by binding the active site of NS5B. They must be phosphorylated before being active. Because NS5B is a highly conserved region of the HCV genome, nucleoside inhibitors have similar activity against all genotypes and high genetic barriers to resistance.31 In contrast to nucleoside inhibitors, non-nucleoside inhibitors achieve NS5B inhibition by binding to 1 of the at least 5 allosteric enzyme sites resulting in conformational changes of the protein-inhibiting catalytic activity of polymerase. They have genotype specific activity and potential for rapid selection of resistance. The rapid development of resistant mutants is possible with non-nucleoside inhibitors because they bind distantly from the active site of NS5B and mutations at the non-nucleoside inhibitor binding site may not necessarily lead to impairment of enzyme function.
Due to their distinctive binding sites, different polymerase inhibitors could theoretically be used in combination to avoid development of resistance.31 Several HCV NS5B polymerase inhibitors have demonstrated clinical efficacy and advanced to clinical trials.
(1) Nucleoside analog NS5B Polymerase inhibitors
♦ Valopicitabine
Valopicitabine (NM203) is the oral prodrug of the nucleoside analog 20-C-methyl-cytidine. The maximum effective dose was not defined because dosing was limited by the development of gastrointestinal side effects. The efficacy and safety of valopicitabine alone and in combination with pegylated interferon have been assessed in clinical trials.32 Studies of valopicitabine monotherapy indicated that a dose of 400 to 800 mg was optimal; higher doses were not well tolerated due to significant side effects, specially nausea and vomiting. Doses of 200 mg of valopicitabine were well tolerated in combination with peginterferon and effective. Studies in combination with ribavirin were not done due to increased gastrointestinal side effects. One major phase IIb trial examining the combination of valopicitabine with peginterferon failed to demonstrate improved efficacy over the SOC of peginterferon/ribavirin.32 Side effects requiring dose reductions in the higher dose peginterferon/valopicitabine arms limited efficacy and tolerability of this regimen. On review of the clinical data, the U.S. Food and Drug Administration (FDA) revised the clinical trial of treating naive patients by reducing the dose of valopicitabine from 800 mg to 200 or 400 mg/d. Subsequent analysis by the FDA of the risk-benefit profile from the phase II clinical trial resulted in suspension of clinical development of valopicitabine in the United States.33 Although development of valopicitabine for the treatment of chronic hepatitis C in the United States has ceased, trials in Europe in combination with peginterferon/ribavirin are ongoing. Results of these latter trials will determine the ultimate fate of valopicitabine.
♦ R1626
R1626 is a tri-isobutyl ester prodrug of the nucleoside analog R1479 (40-azidocytidine). It is rapidly converted by esterases in gastrointestinal epithelial cells to R1479, whereby it is phosphorylated and becomes a potent and highly selective terminator of HCV NS5B. R1626 was generally well tolerated up to 3,000 mg twice a day with only mild side effects in 67% of cases. Gastrointestinal symptoms (diarrhea and nausea) were common, especially in the group taking 4,500 mg twice a day. In addition, there was pancytopenia suggesting bone marrow suppression with a dose of 4,500 mg twice a day. For these reasons, future clinical trials were limited to 3,000 mg twice a day or less. Sequence analysis of the entire NS5B coding region revealed no known R1479 resistance mutations (S96T or S96T/N142T) or any other amino acid substitutions compared with the baseline sequence. In mid-2008, end-of-treatment response (ETR; undetectable HCV RNA at 48 weeks of therapy) results were presented from the phase IIa study.34 Eighty-four percent of patients receiving R1626 1,500 mg twice a day with SOC achieve ETR compared with 65% receiving SOC. Pockros et al. showed virological response rates during and 24 weeks after the end of the study (equivalent to SVR).35 The high rate of relapse is likely due to the short period of R1626 dosing (4 weeks). R1626 has been withdrawn from further consideration for future drug development in the treatment of chronic hepatitis C. Two major factors led to this decision by the manufacturer: suspicion for significant bone marrow suppression with even short courses of treatment and lack of superior efficacy with the studied regimens, compared with SOC.
♦ R7128
R7128, a prodrug of PSI-6130, is an oral cytidine nucleoside analog polymerase inhibitor. In dose-response studies, there was an increase in virologic response with increasing dose, over a dose range from 750 mg daily every day to 1,500 mg twice daily. A phase Ib clinical study demonstrated a mean HCV RNA reduction of 2.7 log10 IU/mL when administered as monotherapy at 1,500 mg twice a day for 2 weeks in treatment experienced patients infected with genotype 1.36 In this study no serious adverse events were reported with more adverse events occurring in the placebo group. In a phase IIa study, 81 naive-to-therapy, genotype-1 patients were randomized across 3 cohorts (cohort 1, R7128 500 mg twice a day 1 peginterferon 1 ribavirin (SOC); cohort 2, R7128 1,000 mg twice a day 1 SOC; and cohort 3, R7128 1,500 mg twice a day 1 SOC; 20-25 active/5-6 placebo stratification) and treated for 28 Days.37 Baseline characteristics were similar in each group. Subjects receiving 1,000 mg or 1,500 mg twice a day+SOC achieved a rapid virologic response (HCV RNA <15 IU/mL at week 4) in 88% and 85%, respectively versus 19% for placebo+SOC. No apparent differences in HCV RNA reduction across race/ethnicity with respect to R7218 1,000 or 1,500 mg twice a day dosing. No dose-related changes in terms of safety and tolerability were noted. No serious adverse events were noted and most side effects were mild. Preliminary data (up to day 56 in all) are available in 25 HCV genotype-2 and -3 patients (10 non-responders, 15 relapsers to SOC) receiving R7128 1,500 mg twice a day 1 SOC (n=20) compared with placebo 1 SOC (n=5) for 28 days.38 Plasma HCV RNA levels decreased a mean of 5.0 log10 IU/mL (90% rapid virological response [RVR]) in the treatment group compared with 4.3 log10 IU/mL (60% RVR) in the placebo group. Again, no serious adverse events or dose-related side effects were reported with many side effects attributed to peginterferon (fatigue, malaise, and headache). In early 2009, Pharmasset announced that they and their developing partner, Roche, agreed with the FDA on the final design for a phase IIb trial with R7128.39 This study is anticipated to enroll 400 treatment naive, genotype-1 or genotype-4 HCV-infected patients. The primary efficacy end point will be sustained virological response and will involve 5 arms (24 and 48 weeks of total treatment, 500 mg and 1,000 mg of R7128 twice a day for 8 weeks or 12 weeks, and a control arm of pegylated interferon and ribavirin for 48 weeks). In November 2008, Pharmasset, Roche, and InterMune initiated the INFORM-1 trial to investigate the combination of R7128 with InterMune's protease inhibitor (ITMN-191).
♦ IDX184
In January 2009, Idenix announced that it will study its polymerase inhibitor, IDX184, in HCV genotype-1 treatment-naive patients who will receive 1 of 4 doses (25 to 100 mg once a day). The study will include 10 patients who will receive the study drug and 2 patients who will receive a placebo drug. Unlike first-generation HCV nucleoside inhibitors (valopicitabine, R7128, and R1626), IDX184 is a "liver-targeted" prodrug, which theoretically will provide increased anti-HCV efficacy and safety. Preliminary studies in monkeys have shown 95% hepatic extraction of orally administered IDX184 with low systemic IDX184 and nucleoside metabolite levels.40 In chimpanzees infected with HCV genotype 1, once daily oral administration of 10 mg/kg/d of IDX184 produced mean viral load reduction of 2.3 log10 after 4 days of dosing.41
♦ MK-0608
MK-0608 (Merck) is a potent nucleoside analog in preclinical trials.42
(2) Non-nucleoside analogues
♦ HCV-796
HCV-796 is a non-nucleoside polymerase inhibitor that has demonstrated potent antiviral activity in vitro and in patients with chronic hepatitis C. Monotherapy showed a maximum antiviral effect after 4 days of treatment with a mean HCV RNA reduction of 1.4 log100 IU/mL. During monotherapy HCV-796 resistant variants were rapidly selected. HCV-796 resistance was associated with the C316Y amino acid substitution. The combination of HCV-796 and peginterferon alfa-2b produced a mean viral reduction of 3.3-3.5 log10 IU/mL after 14 days of treatment compared to 1.6 log10 IU/mL with peginterferon alfa-2b alone.43 Due to clinically significant elevations of liver enzymes, HCV-796 clinical development was discontinued in the phase 2 program. Recently, data from several new non-nucleoside polymerase inhibitors were presented.
♦ PF 00868554
The development of filibuvir (PF 00868554) is most advanced.44 Filibuvir showed in monotherapy of patients with chronic HCV genotype 1 infection a dose-dependent inhibition of viral replication, with maximum reductions in HCV RNA ranging from 0.97 to 2.13 log10 IU/mL. During monotherapy, mutations associated with resistance at position 423 rapidly occurred indicating a low resistance barrier of filibuvir. Filibuvir is currently investigated in combination with peginterferon alfa-2a and ribavirin. In treatment naïve patients with HCV genotype 1 infection triple therapy was associated with a rapid virologic response rate of 60-75% while no patient in the placebo arm achieved a rapid virologic response. The most frequently reported adverse events were headache, fatigue, insomnia and nausea.
♦ VCH-916
VCH-916 showed a maximum HCV-RNA decline ranging between 0.2 and 2.5 log10 IU/mL within 14 days of treatment.45 Like HCV-796 and filibuvir HCV variants conferring resistance were selected during the course of dosing with VCH-916 over a 14-day period. Sequencing revealed selection of L419S/M, M423T/V/I, I482L and V494A variants during monotherapy indicating that VCH-916 should be used in combination to maintain viral suppression and prevent emergence of resistance.46
♦ ANA598
ANA598 showed a decline of HCVRNA after 3 days of monotherapy ranging between 0.4 and 3.4 log10 IU/mL.47 ANA598 was combined in vitro with IFN alfa, the HCV NS3/4 protease inhibitor telaprevir, the NS5B nucleoside polymerase inhibitor PSI-6130, and the TLR7 agonist ANA773, respectively. The in vitro combination studies demonstrated additive to synergistic antiviral effects of ANA598 in combination with other anti-HCV agents having distinct mechanisms of action and non-overlapping resistance profiles. The study indicates that combination therapy may produce a greater viral load reduction and potentially delay the emergence of drug resistance in vivo. Further studies are planned with ANA598 in combination with standard of care.47
♦ BI 207127
BI 207127 monotherapy showed an HCV-RNA decline after 5 days ranging between 0.6 and 3.1 log10 IU/mL in patients with chronic hepatitis C genotype 1 infection.27 Similar to ANA598, no increase in HCV RNA levels was observed during short term BI 207127 monotherapy. One patient developed a severe generalized erythema with facial involvement, which resolved within 2 days after discontinuation of BI 207127 and after antihistaminic treatment. All other adverse events were rated "mild" or "moderate" and were apparently not dose-related. Further clinical development of the compound in combination therapy is planned.
♦ VCH-222
For VCH-222 only preliminary efficacy results on the first 4 treatment-naïve patients with chronic HCV genotype 1 infection treated for 3 days are available showing a decline of HCV-RNA ranging between 3.2 and 4.2 log10 IU/mL.48
♦ Others
MK-3281, ABT-072 and ABT-333 are additional non-nucleoside polymerase inhibitors in development for which no results on antiviral activity in patients with chronic hepatitis C are yet available.49 ABT-072 and ABT-333 were shown to have oral bioavailability in rats and dogs, in vitro metabolic stability and low potential for drug interactions predicting favorable pharmacokinetics in humans.
3) Combination therapy
The nucleoside polymerase inhibitor R7128 and the protease inhibitor ITMN-191 showed substantial antiviral activity in patients with chronic hepatitis C. The INFORM-1 trial is the first trial to investigate the combination of a nucleoside polymerase inhibitor and a protease inhibitor in patients with chronic hepatitis C.50 Both compounds have different resistance profiles and thus are good candidates for combination therapy. After 14 days of combination therapy (with yet lower doses for both compounds), a decline of HCV-RNA ranging between 2.9 and 5.0 to log10 IU/mL was observed. One patient had undetectable HCV-RNA. No viral rebound was observed, providing encouraging evidence that combinations of direct acting antiviral agents could produce viral suppression without inevitable selection of resistance. Higher doses are currently evaluated. An important question is whether combination therapy is sufficient for achieving a sustained virologic response and what treatment duration would be necessary.
3. Host cell targeting inhibitors
A unique aspect of HCV that has not been observed in other viruses is that the entire viral life cycle is associated with cholesterol metabolism in host cells. Thus, drugs that target cholesterol metabolism might be useful for treating HCV infection.51 Also, drugs targeting the host proteins required for HCV infection, nuclear receptor or anti-receptor antibodies may be more helpful in combating the viral infection.52 Targeting host cofactors of the HCV life cycle by different strategies (inhibition of viral entry, targeting host metabolism, nuclear receptors and other principles) may be a novel rational option, especially because they impose higher genetic barriers for resistance than direct antiviral compounds. However, the principle drawback of these strategies is the greater potential for cellular toxicity.
1) Inhibition of viral entry
(1) Anti-receptor antibodies
Entry into the host cell is the primary step in the HCV life cycle, which makes it an attractive target for antiviral therapies. Attachment and cell entry of HCV is pH dependent and is a clathrin-dependent endocytic pathway.53,54 Although the molecular details regarding how this virus enters a cell are unknown, CD8155 and scavenger receptor class B type 156 seem to be the key receptor components that mediate viral entry. However, other potential receptors play a role in entry of HCV such as low density lipoprotein (LDL) receptor,57 negatively charged glycosaminoglycans, and recently, Evans et al.58 added another molecule to the list of HCV receptors, namely, the tight junction protein claudin-1 (CLDN1). Targeting viral receptors can be accomplished by various methods, including the design of small molecules that bind to proteins and prevent interaction(s) with HCV. The crystal structure of CD81 long extracellular loop enabled the design of small molecules that bind CD81 and prevent its association with HCV E2.59 A recent presentation by Liu et al.60 identified compounds that displayed a dose-dependent inhibition of HCV infection.
(2) Scavenger receptor BI (SR-BI)
Recently, a novel function of SR-Bs for viral antigen uptake and recognition has been suggested; SR-BI may represent a cell-surface receptor for the recognition of viral antigens and be implicated in trafficking exogenous viral antigens toward the major histocompatibility complex (MHC) class I presentation pathway. The SR-BI viral antigen interaction may represent a novel target for therapeutic or preventive strategies aiming at the induction of efficient antiviral immune responses61 as an alternative to the development of anti-HCV antibodies, one could consider anti-SR-B1 human MAbs or anti-CD-81 capable of interfering with HCV infection as potential therapeutic leads. Recent data show that BLT-4 and other inhibitors of SR-BI-mediated lipid transfer not only inhibit HCV entry but also fully restore the potency of neutralizing antibodies in infection assays conducted in the presence of HS/HDL, indicating an intriguing link between neutralization efficiency and stimulation of cell entry.62,63 However, it is too early to know whether the potential for vaccines and passive immunotherapy will be realized. Cholesterol-lowering drugs may be beneficial in patients with chronic hepatitis C by exerting effects on cholesterol metabolism and lipoprotein trafficking via SR-BI.
(3) CD81
Meuleman et al.52 showed that CD81 is a critical receptor for HCV infection in vivo recently. Prophylactic injection of monoclonal anti-CD81 antibodies prevented infection of human liver-uPA-SCID mice, however once an infection occurred, no significant difference in viremia was observed between anti-CD81-treated and control animals (irrelevant antibody). These results strongly support the use of CD81 as a clinical target for HCV prevention, especially in the context of orthotopic liver transplantation.52
2) Targeting host metabolism
HCV seems to be not only an infectious hepatotropic virus but also a metabolic disease64 with a wide area of metabolic disarrangement, including lipid metabolism,65 glucose metabolism66 and vitamin D metabolism.67,68
(1) Host lipid biosynthesis inhibitors
Recently, using the new fluxomic techniques, studies revealed that viral infection takes control of cellular metabolism and drives, among other things, marked increases in fatty acid synthesis. Interfering with glucose-to-fatty acid metabolism could stop viral replication, because fatty acid biosynthesis is not essential in adult humans. It does appear, however, to be essential to the ability of HCV to build their envelopes, reproduce and spread. So, targeting of host lipid metabolism by the existing anti-obesity drugs may represent a new way to block these metabolic changes and inhibit viral replication, and may therefore be a potential novel approach that could improve response rates to treatment.69 While statins have shown promise in vitro, they do not appear to have antiviral activity in vivo at clinically used doses. However, data suggesting synergism with α-IFN, support 'proof-of-concept' for trials combining fluvastatin with standard pegylated interferon plus/minus ribavirin. Cautious, prospective and randomized trials are needed before we can call statin therapy an adjuvant treatment panacea.69 Another class of drugs designed for treating hypercholesterolemia blocks the assembly and secretion of very low density lipoprotein (VLDL). These drugs may also be effective in treating HCV infection because they inhibit release of HCV particles from infected cells.70 In this regard, antisense RNA drugs targeting apoB71 and several microsomal triglyceride protein (MTP) inhibitors72,73 have already been tested in clinical trials because of their ability to block VLDL secretion, thereby lowering the plasma levels of VLDL triglycerides and LDL cholesterol. Long-term treatment with MTP inhibitors led to the toxic accumulation of fat in livers,72,73 thus hampering the approval of these drugs for the treatment of hypercholesterolemia on a long-term basis. However, short-term treatment (up to several weeks) reduced the plasma level of VLDL with only minor adverse effects, which disappeared after drug discontinuation.72 It will be interesting to examine whether short-term treatment with MTP inhibitors is beneficial in treating HCV infection. Of note, the natural flavonoid naringenin, demonstrated an ability to block both VLDL secretion and HCV secretion in paralle.74
(2) Cyclophilin B inhibitors
Another host cell factor involved in HCV RNA replication is the human protein cyclophilin B protein which interacts with the C-terminal region of NS5B and appears to stimulate its RNA binding activity.75 The cyclophilin B inhibitor Debio-025 potently suppresses genotype 1 HCV replication in vivo.76
(3) Insulin resistance
Insulin resistance emerges as a very important host factor in patients with CHC, mainly because it has been related to steatosis development, fibrosis progression and non-response to peg-interferon plus ribavirin.77 Insulin resistance is the main pathogenic factor in the development of steatosis in chronic hepatitis C; both viral induced insulin resistance and metabolic insulin resistance could be implicated in the development of steatosis.78 Insulin resistance, calculated by the homeostasis model assessment (HOMA), has been found to be one of the most important host factors related to the impermanence of virological response to combined therapy in chronic hepatitis C patients.79 Recently, obesity has been identified as a modifiable host factor associated with a lower SVR. An elevated body mass index (BMI) is associated with reduced insulin sensitivity and HCV treatment outcome. This observation has led experts to suggest that managing insulin resistance might improve hepatitis treatment outcome and that insulin resistance seems to be a new target in the management of hepatitis C. The rationale of increasing insulin sensitivity in patients with chronic hepatitis C is based on the premise that insulin resistant state directly or indirectly inhibits the antiviral action of IFN-α-β, or increases the viral fitness making it more resistant to therapy, or both.80,81 Intracellular factors dysregulated by HCV and responsible for the insulin resistant phenotype may play promiscuous effects as they are also involved in regulating IFN-α signaling. These factors include some members of the suppressor of cytokine signaling (SOCS) family82-84 and the protein phosphatase 2A.85 Thus, modulating the levels and/or the activity of these factors may not only reverse hepatic insulin resistance but also help in establishing the IFN-α-induced antiviral state at the site of HCV replication. This is one of the reasons for trying to restore insulin sensitivity in chronic hepatitis C patients, especially those who failed to respond to therapy. However, specific inhibitors of SOCS family members and of the protein phosphatase 2A are either not suitable for in vivo administration or are toxic. Alternatively, increasing insulin sensitivity may be achieved by modulating serum levels of specific cytokines, such as tumor necrosis factor-α (TNF-α), associated with insulin resistance,86,87 but the administration of anti-TNF-α antibodies to chronic hepatitis C patients may be risky.88 Recently, metformin-based triple therapy has been shown to be safe, improving insulin sensitivity and increasing SVR rate by 10% in patients with hepatitis C genotype 1 and insulin resistance (HOMA > 2). This therapy was especially effective in females in whom metformin significantly raised the SVR rate.89
3) NUCLEAR RECEPTORS
(1) Peroxisome proliferator-activated receptor (PPAR)
The PPARs are nuclear factors (amongst others) involved in the regulation of glucose homeostasis. In addition to the direct effects on factors involved in lipid and glucose homeostasis,90-94 PPARs may have insulin sensitizing effects via their anti-inflammatory activity.95,96 Thus, treatment with PPAR agonists results in improved insulin sensitivity via diverse mechanisms, both direct and indirect, and both at the level of the liver and at the level of extrahepatic tissues.90 Also, in a recent study, the level of PPARα mRNA was found to be profoundly suppressed in the liver of chronic hepatitis C patients (about 85% compared to control livers).97 The suppression of PPAR-α leads to the upregulation of nuclear factor (NF)-κB. NF-κB has been shown to accelerate virus replication,98 and it has been speculated that activation of PPAR-α with subsequent NF-κB suppression leads to decreased HCV replication in hepatocytes.99 Given the availability of potent agonists, PPARs may represent a novel pharmacological target in the treatment of liver lesions observed in chronic hepatitis C.
(2) Farnesoid X receptor (FXR)
The bile acid receptors were found to play a role in bile acid-mediated promotion of HCV replication.100 These data suggest a novel mechanism for bile acid-mediated gene regulation at virus and host levels. Importantly, these data may contribute to the finding of better regimens for the treatment of chronic HCV infections by including agents altering the bile acid-mediated FXR pathway.100
(3) Estrogen receptor (ESR)
ESR belongs to the steroid hormone receptor family of the nuclear receptor super family. There are two different forms of the estrogen receptor, usually referred to as α and β, each encoded by a separate gene.101 The novel role of ESR α in regulation of HCV replication has been recently reported.102 Tamoxifen and other anti-estrogens suppress genome replication, as part of ESR resides on the endoplasmic reticulum and interacts with HCV RNA polymerase NS5B, so ESR is suggested to serve as a potential novel target for anti-HCV therapies.102
4) Other
Nitazoxanide
Nitazoxanide is an oral prodrug of a thiazolide (tizoxanide), and was approved for the treatment of protozoal infections.103 In addition to having antiprotozoal and antibacterial activity, nitazoxanide coincidently was discovered to inhibit HCV replication104 through a recently identified host-mediated mechanism of action. Recent studies suggest that nitazoxanide and other thiazolides selectively induce PKR phosphorylation, which leads to increased cell concentration of phosphorylated eIF2, a naturally occurring antiviral intracellular protein.105 This mechanism of action is only triggered when a cell is infected with HCV while nitazoxanide has no effect in uninfected cells, which provides a possible explanation for its very low rate of toxicity. Furthermore, nitazoxanide does not appear to induce antiviral resistance, based on an attempt to produce a resistance to nitazoxanide and tizoxanide in HCV replicon-containing cell lines.106 The combination of nitazoxanide, peginterferon α-2a, and ribavirin increased the percentage of patients with rapid and sustained virologic responses, compared with patients given peginterferon plus ribavirin, without an increase in adverse events.107 Nitazoxanide, a novel protein kinase inducer, has the potential not only to increase the SVR rate but also potentially to shorten the duration of therapy.
CONCLUSIONS
The knowledge acquired regarding the HCV life cycle during the last decade has been enormous and has lead to multiple new anti-HCV therapies under investigation. One anticipates that these oral STAT-C drugs will initially be added to current PEG-RBV therapy, leading to improved response rates and shorter duration of therapy. Future trials will need to evaluate the use of "cocktails" of oral agents to see if viral resistance can be minimized (similar to HIV and HBV). This may potentially be achieved either by a combination of two or more specific inhibitors with non-overlapping resistance profiles such as protease inhibitors with nucleoside and/or non-nucleoside inhibitors or the combination of HCV specific inhibitors with non-HCV specific inhibitors such as cyclophilin inhibitors. In the meantime, we have developed improved strategies for maximizing response rates with current therapy and look to use individual viral kinetic responses to tailor treatment duration.
Acknowledgements
None of the authors have any conflict of interest for this manuscript.
Abbreviations
HCV
hepatitis C virus
IFN
interferon
SVR
sustained virological response
NTR
non-translated region
TLR
toll-like receptor
RIG
retionic acid-inducible gene
MDA
melanoma differentiation-associated gene
DC
dendritic cells
STAT-C
specifically targeted therapy against hepatitis C
IRES
internal ribosome entry site
siRNA
small interfering RNAs
SOC
the standard of care
FDA
the U.S. Food and Drug Administration
ETR
end-of-treatment response
RVR
rapid virological response
LDL
low density lipoprotein
MHC
major histocompatibility complex
VLDL
very low density lipoprotein
MTP
microsomal triglyceride protein
HOMA
homeostasis model assessment
BMI
body mass index
SOCS
suppressor of cytokine signaling
TNF
tumor necrosis factor
PPAR
Peroxisome proliferator-activated receptor
FXR
Farnesoid X receptor
ESR
Estrogen receptor