Gut-derived lipopolysaccharide promotes alcoholic hepatosteatosis and subsequent hepatocellular carcinoma by stimulating neutrophil extracellular traps through toll-like receptor 4
Article information

Abstract
Background/Aims
Binge drinking leads to many disorders, including alcoholic hepatosteatosis, which is characterized by intrahepatic neutrophil infiltration and increases the risk of hepatocellular carcinoma (HCC). Molecular mechanisms may involve the migration of bacterial metabolites from the gut to the liver and the activation of neutrophil extracellular traps (NETs).
Methods
Serum samples from both binge drinking and alcohol-avoiding patients were analyzed. Mouse models of chronic plus binge alcohol-induced hepatosteatosis and HCC models were used.
Results
A marker of NETs formation, lipopolysaccharide (LPS), was significantly higher in alcoholic hepatosteatosis and HCC patients and mice than in controls. Intrahepatic inflammation markers and HCC-related cytokines were decreased in mice with reduced NET formation due to neutrophil elastase (NE) deletion, and liver-related symptoms of alcohol were also alleviated in NE knockout mice. Removal of intestinal bacteria with antibiotics led to decreases in markers of NETs formation and inflammatory cytokines upon chronic alcohol consumption, and development of alcoholic hepatosteatosis and HCC was also attenuated. These functions were restored upon supplementation with the bacterial product LPS. When mice lacking toll-like receptor 4 (TLR4) received chronic alcohol feeding, intrahepatic markers of NETs formation decreased, and hepatosteatosis and HCC were alleviated.
Conclusions
Formation of NETs following LPS stimulation of TLR4 upon chronic alcohol use leads to increased alcoholic steatosis and subsequent HCC.
Graphical Abstract
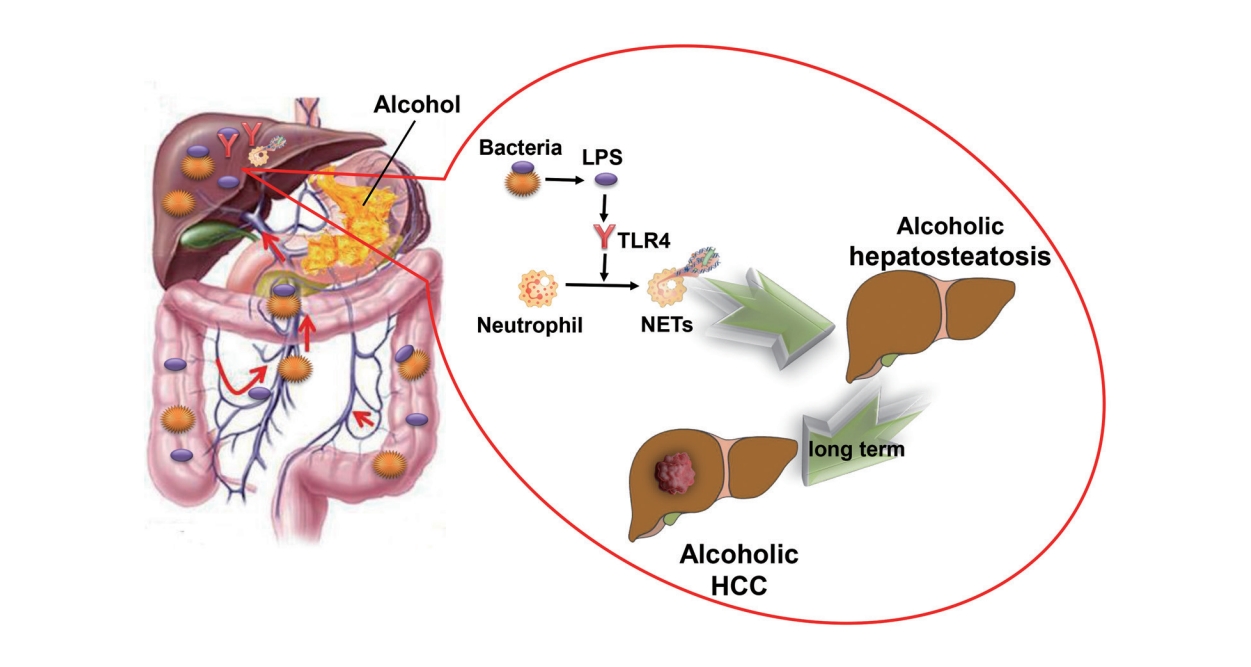
INTRODUCTION
Alcohol consumption has emerged as a leading health risk factor impacting people around the world. According to an annual report of the World Health Organization (WHO), although the global percentage of drinkers has declined, the total alcohol consumption per capita has steadily increased since 2000 [1]. In 2018, the worldwide annual average alcohol consumption was equivalent of 6.4 L of pure alcohol per person; when considering only the people classified as “drinkers,” the average consumption increased to 15.1 L. Importantly, 39.5% of drinkers reported heavy episodic drinking [1].
These increasing numbers are noteworthy in part because chronic alcohol use can lead to many diseases. The most common disease associated with alcohol is hepatosteatosis, which, in the long term, increases the incidence of hepatocellular carcinoma (HCC) [2,3]. Abuse of alcohol, defined as an average daily alcohol consumption of over 80 g, contributes to 15.7% of liver cancer in China [4]. In the United States, which is an area of low hepatitis B virus (HBV) prevalence, it has been reported that 32% of HCC cases are related to alcohol consumption [5]. As HBV vaccination becomes more prevalent and examination of blood used for transfusion becomes more effective, it can be anticipated that the proportion of HCC related to hepatic viruses will drop, and alcohol will eventually become the leading cause of HCC [6]. Therefore, understanding the etiology of HCC, especially in the context of alcohol consumption, is important for its prevention and treatment.
Interestingly, binge drinking is associated with an accumulation of neutrophils in the liver, and this accumulation is a hallmark of alcohol consumption [7]. In addition, neutrophil extracellular traps (NETs), which could be formed after the stimulation of lipopolysaccharide (LPS) both in vivo and in vitro, could contribute to liver injury after binge alcohol use [8-10]. NETs are the networks of extracellular fibers, consisting of neutrophil DNA, myeloperoxidase (MPO), neutrophil elastase (NE) and citrullinated histone, that form in the presence of exogenous copper [11,12]. While NETs are important in the removal of pathogens, NETs may sometimes also damage adjacent cells [13]. Studies have shown that the inhibition of neutrophils can prevent alcoholic steatohepatitis as well as HCC that is induced by chemical carcinogens [14,15]. For example, Kolaczkowska et al. [16] showed that damage was significantly decreased when NETs were depleted in the context of bacterialinduced liver injury. Thus, neutrophils and NETs appear to play a critical role in several aspects of liver damage. Whether these factors also contribute to alcoholic hepatosteatosis and alcohol-related HCC remains unclear.
It is well-known that alcohol contributes to hepatosteatosis and HCC through direct insult to the liver [3]; however, beyond this direct mechanism, alcohol may also damage the intestinal barrier and induce overgrowth of gut bacteria [17]. As a result, overgrowth of bacteria and overproduction of immunogenic components, including LPS, would translocate to the liver, potentially contributing to intrahepatic inflammation, which has been proved to be related to steatosis and carcinogenesis [17-19]. Although LPS has been proved to trigger NETs formation in many conditions, binge alcohol suppressed additional LPS-induced hepatic NET formation in mice [8]. Whether intestine-derived LPS is essential for the formation of NETs in alcoholic hepatosteatosis and subsequent HCC still remains uncertain. According to the evidence presented above, we hypothesized that NETs and LPS play key roles in alcoholic hepatosteatosis and alcohol-induced HCC, and performed the following study to test this hypothesis.
MATERIALS AND METHODS
Collection of human samples
Serum samples were collected from patients who were hospitalized in the Liver Disease Center or General Surgery Department at the Second Affiliated Hospital of Xi’an Jiaotong University. Alcoholic liver disease was diagnosed based on the guidelines issued by the Chinese Society of Gastroenterology [20], and all of the HCC patients were diagnosed by histology. Control serum samples were collected from patients who were diagnosed with inguinal hernia, gallstones, and thyroid nodular goiter and without histories of alcohol consumption or any liver disease prior to their surgeries at the surgical clinic of the General Surgery Department, Second Affiliated Hospital of the Xi’an Jiaotong University. All human sample collections and animal experiment were reviewed and approved by the Ethics Committee of Second Affiliated Hospital of the Xi’an Jiaotong University (No. 2018-2115).
Animals
Wild type (WT) C57BL/6 mice were obtained from the Experimental Animal Center of Xi’an Jiaotong University School of Medicine. NE (#006112), and toll-like receptor (TLR) 4 knockout (KO) mice (#007227) (C57BL/6 background) were obtained from the Jackson Laboratory. Mice were maintained under 12-hour light-dark cycles in specific-pathogen free rooms of the Experimental Animal Center, Xi’an Jiaotong University School of Medicine.
Animal models
A mouse model of chronic plus binge feeding alcoholic hepatosteatosis was established by feeding male mice (8–10 weeks of age) a Lieber-DeCarli liquid alcohol diet (Trophic Animal Feed High-Tech Co., Nantong, China) for 4 weeks followed by three gavages of a single dose of ethanol (5 g/kg body weight per day). Control mice were fed with a Lieber-DeCarli regular control diet and saline. All mice were euthanized at 9 hours after the last gavage for sample harvest.
An animal model of alcoholic HCC was also established. Since HCC does not arise solely through the use of alcohol [21], mice were also treated with diethylnitrosamine (DEN) (Cool Chemistry, Hefei, China; Cat. #55-18-5) to induce HCC. Male mice (4 weeks of age) were intraperitoneally injected with DEN (75 mg/kg) once a week from weeks 4 through 6. From weeks 7 through 9, the dose was increased to 100 mg/kg once a week. Mice were fed a 4% Lieber-DeCarli liquid alcohol diet from weeks 10 to 17, and they were euthanized for sample harvest after week 17. Control mice were treated with the same protocol, except alcohol feeding. LPS (Sigma, St. Louis, MO, USA; Cat. #L5293) solution in phosphate-buffered saline (PBS) was administered daily by subcutaneous injection (300 μg/kg/d).
A mouse model with depleted intestinal microbiota was established by treatment with a cocktail of antibiotics. The following antibiotics were dissolved in drinking water or in a liquid diet: neomycin (1 g/L) (Aladdin, Shanghai, China; Cat. #N109017), ampicillin (1 g/L) (Aladdin; Cat. #A102048), metronidazole (1 g/L) (Aladdin; Cat. #M109874), and vancomycin (0.5 g/L) (Aladdin; Cat. #V105495). To establish the model, the mice were treated with this cocktail for 1 week. Stool was collected and tested by plating on blood agar and counting colony forming units to quantify the depletion of bacteria.
Immunofluorescence
Fresh tissue was fixed in 4% paraformaldehyde overnight. Fixed tissues were dehydrated with a series of alcohol solutions, and the tissue was embedded into paraffin blocks. After slices were prepared, paraffin was removed with xylene, and the slice was hydrated with an ethanol gradient. Antigen was unmasked with sodium citrate and a methanol incubation. Non-specific binding sites were blocked with goat serum (Beijing Solarbio, Beijing, China; Cat. #SL038), and the tissues were incubated with primary antibody (citrullinated histone H3 [Cit H3]; Abcam, Cambridge, UK; Cat. #ab5103; NE, Bioss, Beijing, China; Cat. #bs6982-r) overnight at 4°C. After several washes, samples were incubated with a fluorescent dye-labeled secondary antibody for 1 hour, and nuclei were counterstained with DAPI (Beyotime, Shanghai, China; Cat. #C1002). Slides were mounted with antifade mounting medium. The fluorescence intensity measurement was calculated from at least three adjacent sections using the ImageJ software (v1.8.0, National Institutes of Health, Bethesda, MD, USA).
Quantitative real time-polymerase chain reaction (PCR)
Fresh tissues were homogenized in Trizol solution (Sangon Biotech, Shanghai, China; Cat. #B511311). Then, RNA was extracted according to the manufacturer’s instructions. RNA was diluted with RNA-free water, and DNA was removed with the TURBO DNA-free Kit (ThermoFisher, Waltham, MA, USA; Cat. #AM1907) according to the manufacturer’s protocol. RNA concentrations were determined with a NanoDrop 2000. RNA (1 μg) was used to make cDNA with cDNA Synthesis SuperMix (Bioteke, Shanghai, China; Cat. #PR6502). A final quantitative PCR (qPCR) 20-μL reaction mixture was made with 1-μL cDNA, 10-μL 2X SYBR GREEN MasterMix (Bioteke; Cat. #PR1702), and water, and qPCR was performed on an Exicycler™ 96 (Bioneer, Daejeon, Korea) machine. Primer sequences are shown in Supplementary Table 1. GAPDH was used as a control gene. Relative mRNA expression was calculated by the 2-△△ cycle threshold method with the target gene expression in the sham treatment WT mice group set as 1.
Determination of serum LPS concentration
Serum LPS concentrations were measured with a chromogenic LPS detection kit (Xiamen Bioendo, Xiamen, China; Cat. #EC32545S) according to the manufacturer’s instructions. In brief, 50 μL of diluted serum samples (1:100) were incubated with 50 μL of a limulus amebocyte lysate for 12 minutes at 37°C. Then, the mixture was incubated with 100 μL of the chromogenic substrate solution for 6 minutes at 37°C. Absorbance (λ=410 nm) was measured after the addition of 100 μL of acetic acid.
Statistical analysis
SPSS ver. 20.0 software (IBM, Armonk, NY, USA) was used for data analysis. Numerical data are shown as mean±standard deviation. Student’s t-tests were used to compare the differences between groups. Pearson correlation method was used to perform correlation analysis between serum LPS and MPO-DNA. P-values less than 0.05 were considered significantly different.
Detailed methodology is described in Supplementary Methods.
RESULTS
NETs and inflammation increased in human alcoholic liver disease (ALD) and alcoholic HCC
Serum samples were collected from a total of 42 patients ALD. The characteristics of patients are shown in Table 1, and some of the parameters were different between the control and ALD patients, HCC patients with or without ALD, which imply that liver injury was present in patients with ALD. Based on our study results, ALD patients had a higher serum concentration of MPO-DNA complexes compared to the normal control patients (P<0.05) (Fig. 1A). Nonetheless, no significant difference was found after the subgroup analysis of MPO-DNA level according to liver cirrhosis or tumor stage (Supplementary Fig. 1). Meanwhile, serum concentrations of LPS and interleukin (IL)-6 were also higher in ALD patients (Fig. 1B, C). Serum samples from 36 HCC patients without ALD and 31 patients with both ALD and HCC were collected. The characteristics of patients are shown in Table 1. The serum concentration of MPO-DNA was significantly higher in ALD plus HCC patients than in HCC patients without ALD (P<0.05) (Fig. 1A). Meanwhile, the serum LPS and IL-6 concentrations were significantly higher in ALD plus HCC patients than in HCC without ALD patients (P<0.05) (Fig. 1B, C). Interestingly, there was a significant correlation between serum LPS and MPO-DNA in both ALD patients and ALD plus HCC patients (P<0.05) (Fig. 1D, E).
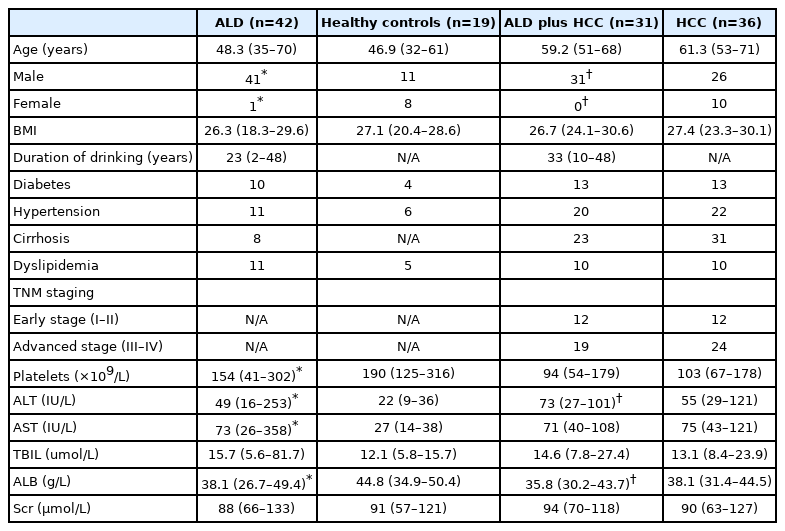
Characteristics of patients with and without ALD

Alcohol consumption increases neutrophil extracellular traps (NETs) in both human and mice. (A-C) Serum MPO-DNA, lipopolysaccharide (LPS), and interleukin (IL)-6 levels from alcoholic liver disease (ALD), hepatocellular carcinoma (HCC), ALD+HCC, and control patients were detected by ELISA. (D, E) Correlation between serum LPS and MPO-DNA in patients with ALD and ALD+HCC. (F) Liver MPO-DNA levels in the mouse models of alcoholic hepatosteatosis and HCC were detected by ELISA. (G, H) Mouse livers were tested for NETs by detecting the expression of citrullinated histone H3 (Cit H3) and neutrophil elastase (NE) by immunofluorescence in both alcoholic hepatosteatosis and alcoholic HCC models (scale bar, 50 μm). OD, optical density; DAPI, 4’,6-diamidino-2-phenylindole; EtOH, ethanol; DEN, diethylnitrosamine; KO, knockout. *P<0.05 compared between groups (5–8 mice per experimental group).
Alcohol increases the formation of intrahepatic NETs in mice
Neutrophil infiltration is a typical pathologic feature of alcoholic liver disease [22], and it could be also seen in a murine model of chronic-binge alcohol consumption [23]. Here, we found that chronic-binge alcohol consumption increased liver MPO-DNA in both alcoholic hepatosteatosis and alcoholic HCC models (Fig. 1F). We then investigated the levels of other markers of NET formation, Cit H3 and NE, by immunofluorescence. This assay indicated more fluorescence in the mice of alcoholic hepatosteatosis models compared to the paired mice with control diets (Fig. 1G, H). The strength of fluorescence was also significantly increased in alcoholic hepatosteatosis groups compared to pair feeding groups (P<0.05) (Fig. 1G, H). The fluorescence DEN plus alcohol treatment group was significantly higher than the fluorescence in only DEN treatment group (P<0.05) (Fig. 1G, H).
NETs promote alcohol-induced hepatosteatosis and alcohol-related HCC in mice
Since NE plays a critical role in lysing of neutrophil membranes in the formation of NETs, NETs do not form in NE KO mice. These mice, then, provided an important model to test the impact of NETs on the development of alcohol-related disorders. In the present study, we found that much less MPO-DNA and Cit H3 were observed in NE KO mice compared to WT mice in the model of alcohol-induced hepatosteatosis and alcohol-related HCC (Fig. 1F, G). After 5 weeks of alcohol consumption, positive regions stained by oil red, which stains intrahepatic fat, were significantly less in NE KO mice than in WT mice treated similarly (Fig. 2A). Liver triglyceride (TG) increased in binge alcohol-treated mice, but this increase in liver TG was attenuated in NE KO mice (Fig. 2B). Serum alanine aminotransferase (ALT) was elevated significantly in alcohol-fed mice as compared to paired controls, and this increase was attenuated in NE KO mice (P<0.05) (Fig. 2C). Similarly, while liver mRNA expression of the inflammation markers tumor necrosis factor (TNF)-α, IL-6, cyclin B1, C-X-C motif ligand 2 (CXCL2), and chemokine ligand 2 (CCL2) were increased upon binge alcohol administration for 5 weeks, these increases were significantly lower in NE KO mice (P<0.05) (Fig. 2D).
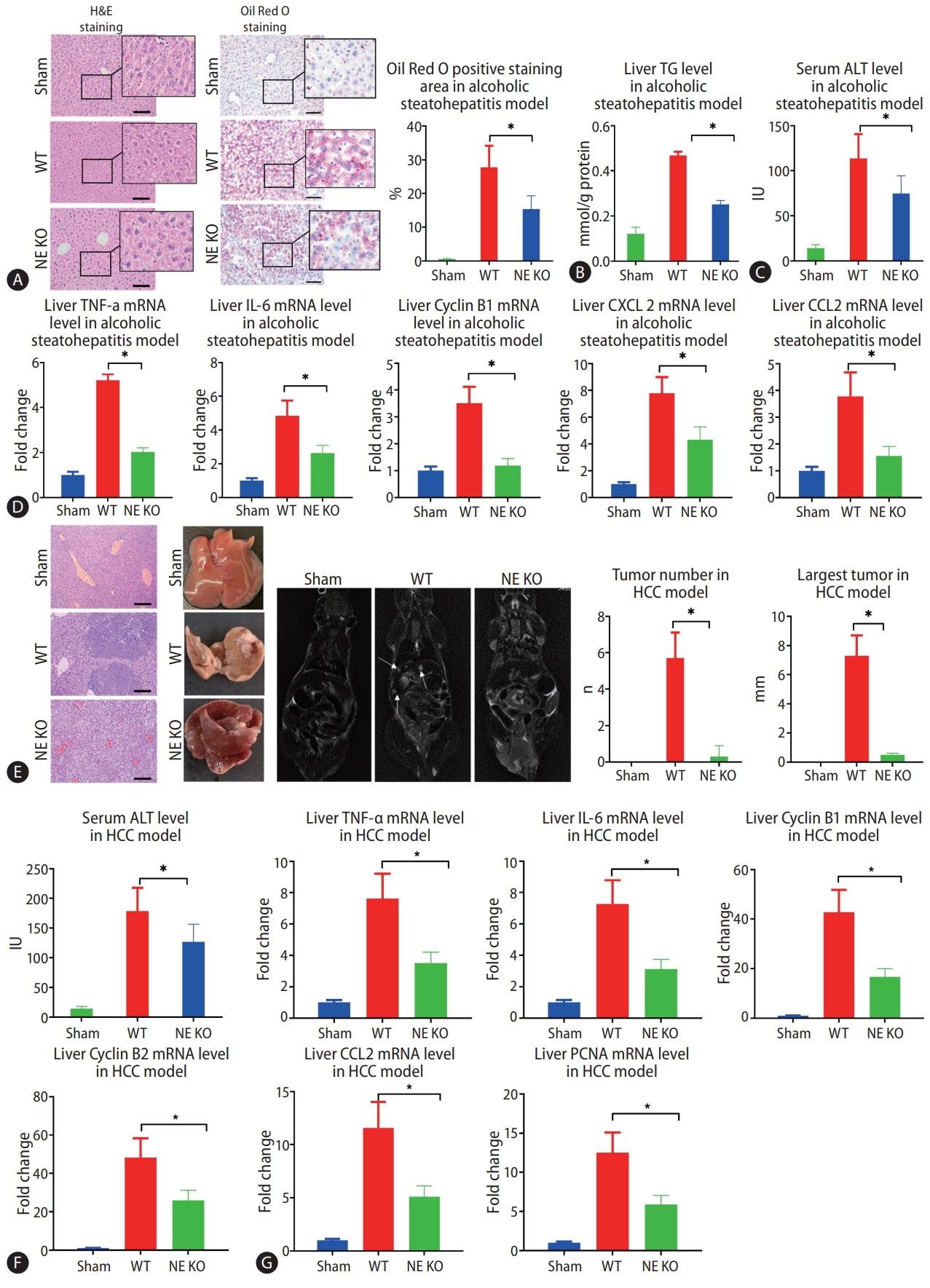
Lack of neutrophil extracellular traps (NETs) leads to alleviation of alcoholic hepatosteatosis and subsequent hepatocellular carcinoma (HCC) in mice. (A) Mouse liver tissues were visualized by hematoxylin and eosin (H&E) staining and Oil Red O staining after 5 weeks of alcohol treatment (scale bar, 100 μm). (B, C) Liver triglyceride (TG) and serum alanine aminotransferase (ALT) levels from alcoholic hepatosteatosis model mice were determined. (D) Wild type (WT) and neutrophil elastase (NE) knockout (KO) mice underwent alcohol treatment for 5 weeks, and liver levels of tumor necrosis factor (TNF)-α, interleukin (IL)-6, cyclin B1, C-X-C motif ligand 2 (CXCL-2), and chemokine ligand 2 (CCL2) genes were determined by qPCR. (E) Liver tumors from HCC model mice were analyzed by H&E staining (scale bar, 200 μm), photography, and magnetic resonance imaging (MRI) scanning. The arrows indicate the location of tumors in MRI scanning. (F) Serum ALT levels were determined in mouse model of HCC. (G) WT and NE KO mice underwent the procedure to establish the HCC model for 13 weeks, whereupon liver levels of TNF-α, IL-6, cyclin B1, Cyclin B2, CCL2, and proliferating cell nuclear antigen (PCNA) genes were determined by qPCR. qPCR, quantitative polymerase chain reaction. *P<0.05 compared between groups (5–8 mice per experimental group).
In the murine model of alcoholic HCC, some tumors were formed in WT mice, but few tumors were seen in NE KO mice. Both average tumor number and maximum tumor size were significantly higher in WT mice relative to NE KO mice (P<0.05) (Fig. 2E). Meanwhile, serum ALT concentrations were decreased in NE KO mice as compared to WT mice (Fig. 2F). We further tested the liver mRNA levels of CCL2, cyclin B1, cyclin B2, IL-6, TNF-α and proliferating cell nuclear antigen (PCNA), and results showed that mRNA expression of all of these genes was lower in NE KO mice than WT mice (P<0.05) (Fig. 2G).
Intestinal bacteria and LPS promote NETs formation in the mice liver upon chronic alcohol consumption
After we confirmed that alcohol stimulated the formation of intrahepatic NETs and further relation of this increase to alcohol-induced hepatosteatosis and HCC, we endeavored to figure out an upstream inducer of NET formation. Since the gut-liver axis has been shown to play a critical role in many liver diseases, we considered that some product of intestinal bacteria could be the key factor. When we depleted gut bacteria with a cocktail of antibiotics in mice of the alcohol-induced hepatosteatosis model, we found that intrahepatic MPO-DNA was significantly decreased (P<0.05) (Fig. 3A). In addition, upon antibiotic treatment, fewer cells staining positive for intrahepatic Cit H3 and NE were observed (Fig. 3B, C) indicating lower NET formation.

Gut-derived lipopolysaccharide (LPS) promotes intrahepatic neutrophil extracellular trap (NET) formation during mice alcoholic hepatosteatosis and subsequent hepatocellular carcinoma (HCC). (A) Liver MPO-DNA was determined by ELISA in the mouse model of alcoholic hepatosteatosis. (B, C) Alcoholic hepatosteatosis livers were tested for NETs according to the expressions of citrullinated histone H3 (Cit H3) and neutrophil elastase (NE), which were determined by immunofluorescence (scale bar, 50 μm). (D) HCC liver MPO-DNA was determined by ELISA in the mouse model of HCC. (E, F) HCC livers were tested for NETs according to the expressions of Cit H3 and NE, which were determined by immunofluorescence (scale bar, 50 μm). OD, optical density; Abx, antibiotics; TLR, toll-like receptor; KO, knockout; DAPI, 4’,6-diamidino2-phenylindole. *P<0.05 compared between groups (5–8 mice per experimental group).
When mice were treated with both antibiotics and LPS, although the bacterial population of stool was significantly reduced, both stool and liver LPS levels were increased (Supplementary Fig. 2A, C). After a 5 weeks binge alcohol treatment, liver MPO-DNA was recovered with the combined treatment, and intrahepatic NE and Cit H3-positive cells also increased relative to mice treated only with antibiotics (Fig. 3A-C).
Similar results were also seen in the alcoholic HCC model. Depletion of intestinal bacteria by a cocktail of antibiotics attenuated intrahepatic LPS (Supplementary Fig. 2), and liver MPO-DNA and intrahepatic cells positive for NE and Cit H3 also significantly decreased (Fig. 3D-F). On the other hand, in this model, antibiotics failed to attenuate liver MPO-DNA level or the fluorescence of NE and Cit H3-positive cells when mice were also treated with LPS (Fig. 3D-F).
LPS plays a role in mice alcoholic hepatosteatosis and alcohol-related HCC
A we have shown that LPS plays a role in the alcohol-induced formation of intrahepatic NETs, and NETs are known to correlate with alcoholic hepatosteatosis and alcohol-related HCC. Therefore, we predicted that LPS contributes to these diseases. Accordingly, in the model of alcohol-induced hepatosteatosis, antibiotics decreased the ballooning degeneration of hepatocytes and inflammation of intrahepatic cells as demonstrated by hematoxylin and eosin (H&E) staining, while these phenomena increased when antibiotic treatment was combined with LPS administration. Similarly, upon staining with Oil Red O, less red staining was observed in the mice treated with antibiotics, but more staining was observed when antibiotics were combined with LPS (Fig. 4A). Next, we determined the concentrations of TG in the liver, and found that it was decreased in the antibiotics group but was recovered in the group treated with antibiotics and LPS (Fig. 4B). Serum ALT levels were much lower in the antibiotics group and were increased with LPS (Fig. 4C). Meanwhile, liver expressions of the mRNAs of TNF-α, cyclin B1, CCL2, and CXCL2 were significantly decreased after treatment with antibiotics, while this effect was attenuated upon treatment with LPS (P<0.05) (Fig. 4D).

Intestinal lipopolysaccharide (LPS) contributes to alcohol-induced hepatosteatosis and subsequent hepatocellular carcinoma (HCC) in mice. (A) Hepatosteatosis was determined by hematoxylin and eosin (H&E) and Oil Red O staining after treatment with alcohol for 5 weeks (scale bar, 100 μm). (B, C) Liver triglyceride (TG) and serum alanine aminotransferase (ALT) concentrations were determined in the mouse model of alcoholic hepatosteatosis. (D) Liver levels of tumor necrosis factor (TNF)-α, interleukin (IL)-6, cyclin B1, C-X-C motif ligand 2 (CXCL-2), and chemokine ligand 2 (CCL2) genes were determined by qPCR in the alcoholic hepatosteatosis model. (E) Mouse liver tumors were presented by photography, H&E staining (scale bar, 200 μm), and magnetic resonance imaging (MRI) scan after development of the HCC model. The arrows indicate the location of tumors in MRI scanning. (F) The serum ALT concentration was determined in HCC mice. (G) Liver levels of TNF-α, IL-6, Cyclin B1, cyclin B2, CCL2, and proliferating cell nuclear antigen (PCNA) genes were determined by qPCR in the HCC model. Abx, antibiotics; qPCR, quantitative polymerase chain reaction. *P<0.05 compared between groups (5–8 mice per experimental group).
In the alcohol-related HCC model, the use of antibiotics attenuated HCC carcinogenesis. The number of tumors and the maximum tumor size were significantly reduced in the mice that were administered antibiotics, while the number and size increased upon combining of antibiotics with LPS treatment (Fig. 4E). At the time of sacrifice, serum ALT concentrations were significantly lower in the antibiotic-treated mice than in the mice not treated with antibiotics, and these concentrations were recovered upon co-administration of LPS (Fig. 4F). Similarly, liver expressions of the mRNA of CCL2, cyclin B1, cyclin B2, TNF-α, IL-6, and PCNA were significantly decreased upon treatment of mice with antibiotics, and the expressions of these genes were increased again upon co-treatment of mice with antibiotics and LPS (Fig. 4G).
Lack of TLR4 decreases NET formation in mice during chronical alcohol consumption
After confirming that intestinal-derived LPS promotes the formation of NETs during chronic alcohol consumption, we considered that TLR4, an LPS receptor, may also play a role in the formation of NETs. A strain of WT and TLR4 KO mice was used in the study. There was no difference in the concentration of intrahepatic LPS between the mice with and without TLR4 in the model of alcohol-induced hepatosteatosis (Supplementary Fig. 2C). However, when we examined the expressions of Cit H3 and NE in the liver by immunofluorescence, we observed fewer positive cells in TLR4 KO mice than in WT mice (Fig. 5A, B). Also, the liver MPO-DNA level decreased significantly in the absence of TLR4 (Fig. 5C).
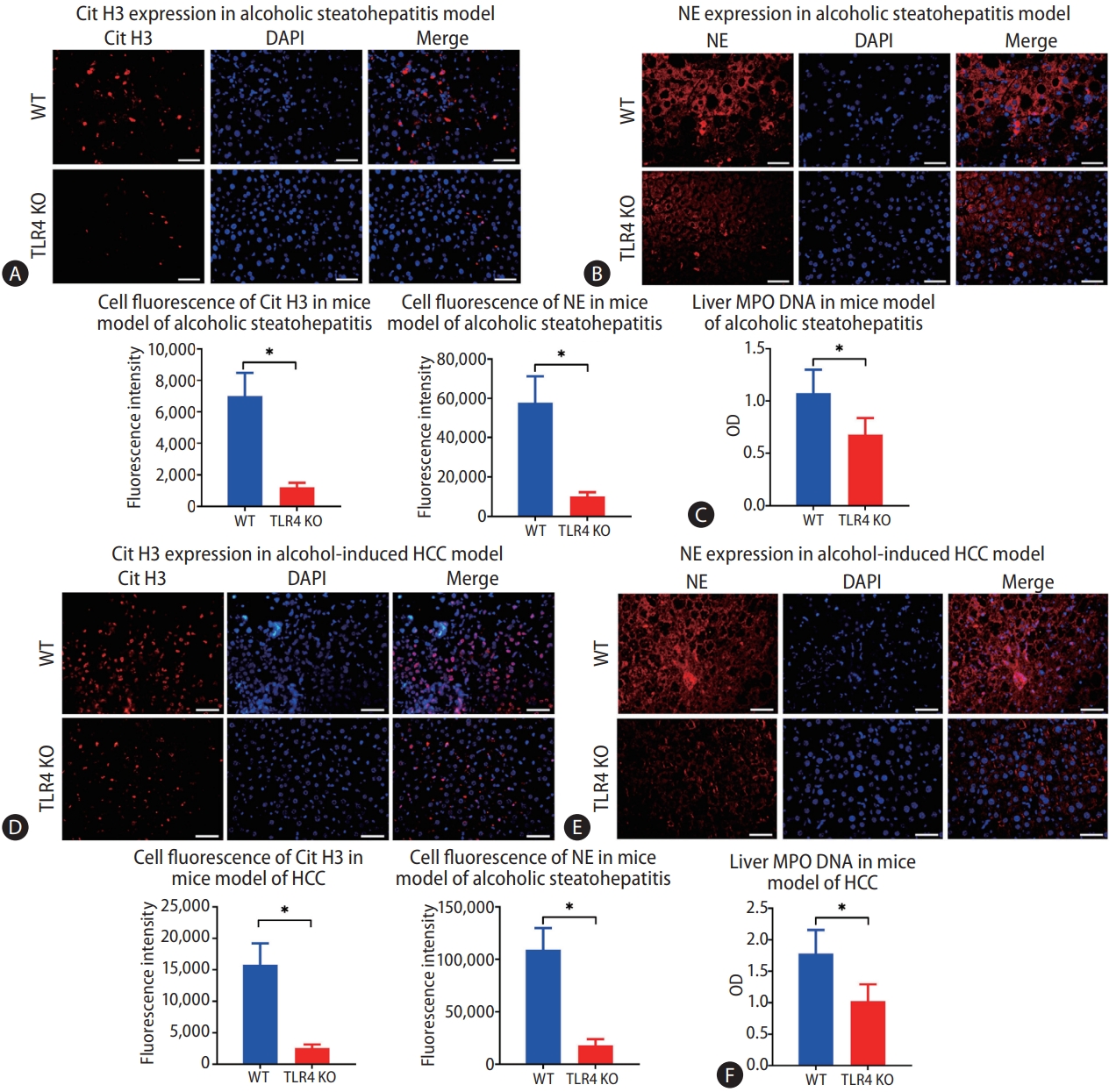
Lack of toll-like receptor 4 (TLR4) results in less intrahepatic neutrophil extracellular trap (NET) formation during the process of alcoholic hepatosteatosis and subsequent hepatocellular carcinoma (HCC) in mice. (A, B) Intrahepatic NETs were determined by immunofluorescence staining of citrullinated histone H3 (Cit H3) and neutrophil elastase (NE) in both wild type (WT) and TLR4 knockout (KO) mice after treatment with alcohol for 5 weeks (scale bar, 50 μm). (C) Liver MPO-DNA was tested by ELISA in both WT and TLR4 KO mice in a model of alcoholic hepatosteatosis. (D, E) Livers from WT and TLR4 KO mice were tested for NETs according to the expressions of Cit H3 and NE, which were determined by immunofluorescence in the HCC model (scale bar, 50 μm). (F) Liver MPO-DNA was determined by ELISA in the HCC model in both WT and TLR4 KO mice. DAPI, 4’,6-diamidino-2-phenylindole. *P<0.05 compared between groups (5–8 mice per experimental group).
The same pattern was observed in the alcohol-related HCC model. The intrahepatic LPS was constant upon depletion of TLR4, but fewer cells were stained positive for Cit H3 and NE in TLR4 KO mice than in WT mice (Fig. 5D, E). Similarly, the liver MPO-DNA concentration was reduced significantly in TLR4 KO mice compared to WT mice (Fig. 5F).
Lack of TLR4 decreases formation of NETs and HCC in the mouse model of alcohol-induced HCC
Finally, we investigated whether TLR4 also plays a role in alcohol-induced hepatosteatosis and alcohol-induced HCC by comparing WT and TLR4 KO mice. In the murine model of alcohol-induced hepatosteatosis, fewer ballooning degeneration hepatocytes and intrahepatic inflammation cells could be seen in the liver by H&E staining. Intrahepatic fat deposition, as revealed by Oil Red O staining, was observed less in TLR4 KO mice than in WT mice (Fig. 6A). Serum ALT and liver TG concentrations were much decreased in TLR4 KO mice (Fig. 6B, C). Liver expressions of CXCL2, CCL2, IL-6, TNF-α, and cyclin B1 mRNA were higher in WT mice than in TLR4 KO mice (Fig. 6D).
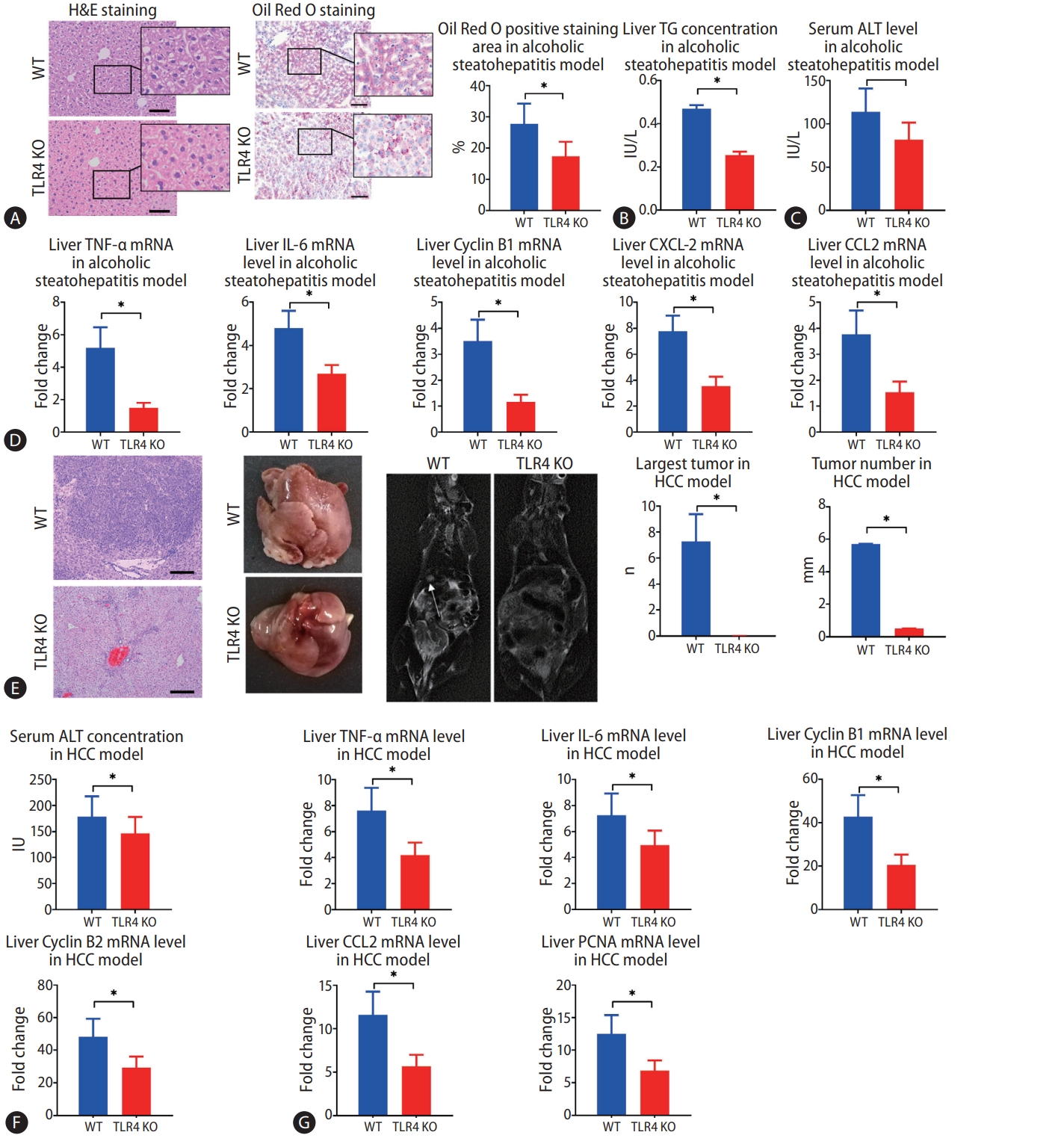
Mice alcoholic hepatosteatosis and subsequent hepatocellular carcinoma (HCC) are attenuated in the absence of toll-like receptor 4 (TLR4). (A) Hepatosteatosis was determined by hematoxylin and eosin (H&E) and Oil Red O staining after treatment with alcohol for 5 weeks in wild type (WT) and TLR4 knockout (KO) mice (scale bar, 100 μm). (B, C) Liver triglyceride (TG) and serum alanine aminotransferase (ALT) concentrations were determined in WT and TLR4 KO mouse models of alcoholic hepatosteatosis. (D) WT and TLR4 KO mouse liver levels of tumor necrosis factor (TNF)-α, interleukin (IL)-6, cyclin B1, C-X-C motif ligand 2 (CXCL-2), and chemokine ligand 2 (CCL2) genes were determined by qPCR in the alcoholic hepatosteatosis model. (E) WT and TLR4 KO mouse liver tumors were presented by photography, HE staining (scale bar, 200 μm), and magnetic resonance imaging (MRI) scanning after development of the HCC model. The arrows indicate the location of tumors in MRI scanning. (F) Serum ALT concentrations were determined in the HCC model in WT and TLR4 KO mice. (G) Liver levels of TNF-α, IL-6, cyclin B1, cyclin B2, CCL-2, and proliferating cell nuclear antigen (PCNA) genes were determined by qPCR in HCC model in WT and TLR4 KO backgrounds. qPCR, quantitative polymerase chain reaction. *P<0.05 compared between groups (5–8 mice per experimental group).
In the murine model of alcoholic HCC, some tumors had occurred by the time of harvest, but few tumors were observed in TLR4 KO mice, and TLR4 KO mice showed significant decreases in both the average number of tumors and the size of largest tumor (Fig. 6E). Upon euthanization, the serum levels of ALT were found to be significantly decreased in TLR4 KO mice compared to WT mice (Fig. 6F). Meanwhile, the mRNA expressions of PCNA, CCL2, TNF-α, IL-6, cyclin B1, and cyclin B2 were decreased in TLR4 KO mice compared to WT mice (Fig. 6G).
DISCUSSION
Binge drinking is a global health problem, and it is well-known that a potential consequence of binge drinking is hepatic steatosis, which can lead to HCC. In fact, many clinical studies have shown that chronic alcohol consumption is an independent risk factor for HCC [5,24], and the WHO has listed alcohol as a risk factor for HCC occurrence [1]. Animal studies have shown that, although HCC does not occur only with long-term alcohol administration, alcohol consumption significantly accelerates the development of HCC and increases tumor load when combined with a chemical inducer [25].
Intrahepatic neutrophil infiltration is a hallmark of binge alcohol consumption, and neutrophils have been proved to play an important role in the development of HCC [14]. Neutrophils are also believed to be a major source of many cytokines, including matrix metalloproteinase-9, IL-10, and CCL2, which are thought to play roles in enhanced tumorigenesis, invasion, and growth. A strong infiltration of neutrophils into the liver is also believed to be independently associated with poorer survival in human HCC [26]. Moreover, it has been reported that neutrophils contribute to ALD [27], and that antibody-mediated depletion of hepatic neutrophils will attenuate HCC [14]. Intestinal bacterial has also been considered to contribute to ALD [28]. However, the mechanisms behind the involvement of intestinal bacteria and neutrophils in alcoholic HCC have not been fully revealed. In particular, NETs are a neutrophil-formed structure that have been proved to form in nonalcoholic steatohepatitis and HCC, and to be involved with metastasis of HCC and many other liver diseases [29,30]. However, as in the case of neutrophils, the precise role of NETs in alcohol-related HCC has not been revealed.
According to our initial data involving human subjects, we found that ALD and alcohol-related HCC correlated with increased serum levels of MPO-DNA complexes, which are indicators of the formation of NETs. However, without liver biopsy samples, we could not confirm whether NETs were formed in the liver or in extrahepatic organs; therefore, a murine study is necessary. With chronic alcohol feeding mouse models, we found an abundance of NETs formed in the liver under the conditions of chronic alcohol consumption.
The process of NETosis, which is a form of programmed cell death, and the elimination of antigens by NETs, reactive oxygen species (ROS), cytokines, and enzymes are released [31]. This release would not only remove the pathogen but also impair the surrounding healthy cells [31]. Kolaczkowska et al. [16] found that intrahepatic neutrophil infiltration is critical for bacterial-induced liver injury, and liver injury decreased 80% when NET formation was inhibited in the liver. Similarly, NETs have been found to contribute to liver injury associated with sepsis that is induced by binge alcohol consumption [8], along with many other liver disorders not associated with alcohol, such as non-alcoholic steatohepatitis, ischemia reperfusion injury, and portal hypertension [31,32].
However, whether NET formation is a risk factor for alcohol-induced HCC remains uncertain. In this study, the NE KO mice, in which neutrophils fail to lyse and form NETs, were used. Using this model, we found that alcoholic hepatosteatosis, together with many inflammation markers, was significantly attenuated upon the loss of NET formation. It is well-known that chronic inflammation is related to carcinogenesis, and hepatosteatosis is also known as a risk factor for HCC. Accordingly, there is no surprise that the absence of NETs delays HCC occurrence.
While we confirmed that NETs are related to alcoholic hepatosteatosis and HCC, the reason for the intrahepatic accumulation of NETs upon alcohol consumption still remained unclear. Notably, alcohol binge consumption not only insults the liver but also affects the intestinal tract. Alcohol and its metabolites can directly injure the intestinal mucosa and contribute to intestinal inflammation, which may lead to an increase in gut permeability [33]. In addition, consuming alcohol can also slow down intestinal movement and result in the overgrowth of intestinal bacteria [34,35]. Together, these factors can lead to the occurrence of leaky gut and the movement of bacteria and bacterial products into the liver via the portal vein [34,35]. The role of the gut-liver axis has been implicated in many liver diseases, including some alcohol-related liver diseases, and LPS has been shown to be a mediator of the communication between the liver and the gut [36,37]. However, with leaky gut in alcohol consumption, there remain some additional bacterial products that might be involved in intrahepatic translocation, such as LPS.
An important task of the innate immune system is to rapidly eliminate bacteria and their metabolites from the circulation. In a mouse model of sepsis, most bacteria were trapped in the liver immediately upon entry into the blood. Subsequently, neutrophil granulocytes were also immediately recruited to the liver [38]. In the present study, by completely removing the intestinal bacteria in a mouse model, we found that intestinal bacteria were the cause of the formation of NETs induced by alcoholic steatohepatitis and subsequent HCC. However, it should be noted that the existing intestinal sterilization methods that eliminate various intestinal bacteria can also eliminate relevant chemical components, such as LPS and lipoteichoic acid (LTA). Therefore, it is possible that one or both of these chemicals influenced NET formation. A relationship between LTA production and NET formation has been demonstrated [39], and other studies have even shown that LTA protects against NET-induced liver damage [9,40]. Similarly, multiple studies have reported that LPS, a pathogen-associated molecular pattern molecule produced by Gram-negative bacteria, can promote the adhesion of neutrophil granulocytes in the hepatic sinusoid and promote NET formation via activation of the TLR4 receptor [9,10]. The LPS-TLR4 pathway has been found to participate in the progress of both non-alcoholic steatohepatitis and HCC, and NET formation has also been shown to be correlated with such chemical-induced HCC [29,41,42].
Therefore, both LTA and LPS were candidates for chemical mediator of the link between intestinal bacteria and NET formation. Upon removal of intestinal bacteria, we found that NET formation was recovered by supplementing with LPS. In addition, the formation of NETs was inhibited in mice lacking TLR4. Together, these results indicate that intestinal LPS is the key factor leading to NET formation during alcohol intake, and it thereby contributes to the development of alcoholic hepatosteatosis and subsequent HCC.
The activation of TLR4 can also stimulate the formation of NETs through non-bacterial factors. For instance, the platelets of patients with ANCA-associated vasculitis can promote the formation of NETs in vitro through TLR4 without the assistance of bacteria [43]. High mobility group box 1 also causes adjacent damage by promoting formation of NETs through TLR4 in liver ischemia-reperfusion injury [13]. Activation of the LPS-TLR4 pathway has been found to be involved in cell damage and an increase in intracellular ROS upon inhalation of alcohol, thus leading to the release of several damage-associated molecular pattern molecules [44]. Although our study clearly demonstrated a role of TLR4 in the formation of NETs upon alcohol consumption via the study of TLR4 KO mice, the impact of TLR4 on cellular NET formation may be direct or indirect, as LPS activation of TLR4 on nearby cells could stimulate the release of cytokines which would stimulate NET formation. The actual mechanism may also involve both direct and indirect pathways, and elucidating the mechanism will be a subject of further research.
Our findings were consistent with the results of several previous studies which showed that antibiotics can prevent the occurrence of ALD and non-alcoholic HCC [45-47]. In the present study, we identified a role of intestinal bacteria in HCC, and antibiotics seemed to prevent alcoholic HCC. However, since chronic antibiotic treatment is associated with multiple negative side effects, it is not suitable for HCC prophylaxis. Instead, our results suggest that targeting the gut-liver axis or the LPS-TLR4 pathway, maintaining the gut barrier, and detoxifying LPS are likely to represent superior prophylactic strategies [48,49].
In conclusion, our findings showed that intestinal-derived LPS stimulates NET formation via the TLR4 pathway and increases intrahepatic inflammation in chronic alcohol consumption, which in turn increases the development of alcoholic hepatosteatosis and subsequent HCC.
Notes
Authors’ contributions
Y.L.: Designed research, conducted experiments, acquired data, analyzed data, wrote the manuscript. S.C., J.W.: Conducted experiments, acquired data, analyzed data, and wrote the manuscript. X.Z.: Designed research, conducted experiments, analyzed data. S.Y., Y.L, M.X., W.Q, H.A., H.L., T.S., J.W.: Acquired data, conducted experiments. G.C.: Designed research, analyzed data, revised the manuscript. All authors revised and approved the manuscript for publication.
Conflicts of Interest
The authors have no conflicts to disclose.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (Grant No. 81602401) and Natural Science Foundation of Shaanxi Province (2020JM404) to Y.L. The work was also supported by the National Natural Science Foundation of China (Grant No.82003998) to S.C. and (Grant No. 81902449) M.X.
SUPPLEMENTAL MATERIAL
Supplementary material is available at Clinical and Molecular Hepatology website (http://www.e-cmh.org).
Sequences of primers that were used in the study
Serum MPO-DNA level in alcoholic liver disease (ALD), hepatocellular carcinoma (HCC), and ALD+HCC patients. Liver MPO-DNA levels in the mouse models of alcoholic hepatosteatosis and HCC were detected by ELISA. (A) Subgroup analysis of the MPODNA level according to whether the patients were with or without liver cirrhosis. (B) Subgroup analysis of the MPO-DNA level according to the tumor stage. OD, optical density.
Stool bacteria and serum lipopolysaccharide (LPS) levels in both alcoholic hepatosteatosis and hepatocellular carcinoma (HCC) mice. (A, B) Stool CFU was tested every week in the mouse models of alcoholic hepatosteatosis and HCC. (C, D) Serum LPS was tested after euthanization in a mouse model of alcoholic hepatosteatosis and HCC model. CFU, colony forming unit; WT, wild type; Abx, antibiotics; NE, neutrophil elastase; KO, knockout; TLR, toll-like receptor. *P<0.05 compared between groups (5–8 mice per experimental group).
Abbreviations
ALD
alcoholic liver disease
ALT
alanine aminotransferase
CCL2
chemokine ligand 2
Cit H3
citrullinated histone H3
CXCL2
C-X-C motif ligand 2
DEN
diethylnitrosamine
H&E
hematoxylin and eosin
HBV
hepatitis B virus
HCC
hepatocellular carcinoma
IL
interleukin
KO
knockout
LPS
lipopolysaccharide
LTA
lipoteichoic acid
MPO
myeloperoxidase
NE
neutrophil elastase
NETs
neutrophil extracellular traps
PBS
phosphate-buffered saline
PCNA
proliferating cell nuclear antigen
qPCR
quantitative polymerase chain reaction
ROS
reactive oxygen species
TG
triglyceride
TLR
toll-like receptor
TNF
tumor necrosis factor
WHO
World Health Organization
WT
wild type
References
Article information Continued
Notes
Study Highlights
• NET formation in chronic alcohol consumption.
• Intestinal-derived LPS stimulates NET formation via the TLR4 pathway in chronic alcohol consumption.
• Intestinal-derived LPS stimulates NET formation, which in turn increases the development of alcoholic hepatosteatosis and subsequent HCC. Ibusa popubliam ponvocu pplicia ceperum, se critabus id curnu escidef frecta ve, C. Uro consuliam es? Nos