From DCPD to NTCP: The long journey towards identifying a functional hepatitis B virus receptor
Article information
Abstract
Hepatitis B virus (HBV) is the prototype of hepatotropic DNA viruses (hepadnaviruses) infecting a wide range of human and non-human hosts. Previous studies with duck hepatitis B virus (DHBV) identified duck carboxypeptidase D (dCPD) as a host specific binding partner for full-length large envelope protein, and p120 as a binding partner for several truncated versions of the large envelope protein. p120 is the P protein of duck glycine decarboxylase (dGLDC) with restricted expression in DHBV infectible tissues. Several lines of evidence suggest the importance of dCPD, and especially p120, in productive DHBV infection, although neither dCPD nor p120 cDNA could confer susceptibility to DHBV infection in any cell line. Recently, sodium taurocholate cotransporting polypeptide (NTCP) has been identified as a binding partner for the N-terminus of HBV large envelope protein. Importantly, knock down and reconstitution experiments unequivocally demonstrated that NTCP is both necessary and sufficient for in vitro infection by HBV and hepatitis delta virus (HDV), an RNA virus using HBV envelope proteins for its transmission. What remains unclear is whether NTCP is the major HBV receptor in vivo. The fact that some HBV patients are homozygous with an NTCP mutation known to abolish its receptor function suggests the existence of NTCP-independent pathways of HBV entry. Also, NTCP very likely mediates just one step of the HBV entry process, with additional co-factors for productive HBV infection still to be discovered. NTCP offers a novel therapeutic target for the control of chronic HBV infection.
INTRODUCTION
An estimated 350 million people worldwide are chronically infected with hepatitis B virus (HBV), which greatly increases their risk to develop liver cirrhosis and hepatocellular carcinoma (HCC). Currently approved therapies for HBV infection are limited and suboptimal. Pegylated interferons are effective in only a third of patients, and may cause serious side effects. Nucleos(t)ide analogues target DNA replication, a late step in HBV lifecycle. They fail to promote hepatitis B e antigen (HBeAg) or hepatitis B surface antigen (HBsAg) seroconversion, measures of sustained virological response. In this regard, HBV protein expression and genome replication require transcription of viral RNAs from covalently closed circular (ccc) DNA template in the nucleus, whose long half-life is a major obstacle for clearance of HBV infection. As HBV enters hepatocytes through cell surface receptors, targeting viral entry could prevent cccDNA formation in the first place. Numerous attempts have been made to identify the enigmatic receptor(s) for HBV, and the journey started with the related duck hepatitis B virus (DHBV).
dCPD as a host-specific DHBV receptor with high affinity for intact L protein
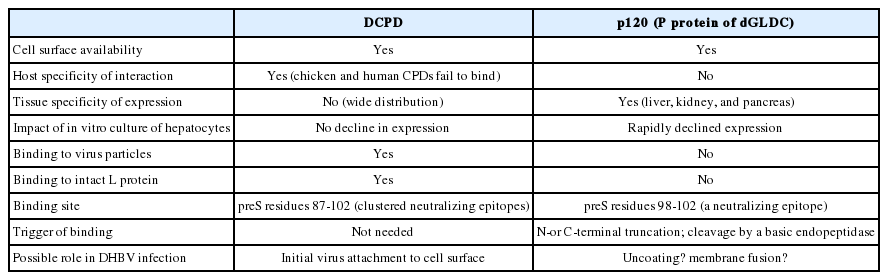
HBV is the prototype of a class of hepatotropic DNA viruses, or hepadnaviridae, which infect the liver (and to a lesser extent, kidney and pancreas) in a host-specific manner. Other members infect ducks, woodchucks, and ground squirrels. DHBV has been the model virus to elucidate key steps in hepadnaviral lifecycle, such as replication by reverse transcription. The low cost of ducks makes it possible to perform extensive in vivo infection experiments, which can be complemented by in vitro infection of primary duck hepatocytes under more defined and controllable conditions. Moreover, as transfection of cloned DHBV DNA into a chicken hepatoma cell line (LMH) leads to DNA replication and virion release, the powerful genetic approach can be employed to define the structural basis for viral infectivity. DHBV produces just two co-terminal envelope proteins through alternative translation initiation, with the large (L) envelope protein having an extra preS domain than the small (S) protein. Evidence suggests that the preS domain mediates high-affinity interaction with the viral receptor, and Dr. Ganem's group identified a 180-kda duck glycoprotein (gp180) as a preS binding partner.1 Two observations made gp180 interesting. First, no binding protein of similar size could be detected from human or chicken tissue suggesting host specificity. Second, gp180 - L protein interaction could be blocked by several neutralizing anti-preS antibodies, but not by a non-neutralizing antibody.1 We independently identified a 170-kda duck glycoprotein (p170) interacting with the preS domain of DHBV L protein.2 Peptide sequencing of p170 and molecular cloning of gp180 revealed them to be the same protein, a trimeric form of basic carboxypeptidase now called duck carboxypeptidase D (dCPD).23 Within the preS domain of 161 residues, we mapped the dCPD binding site to residues 87-102 corresponding to clustered neutralizing epitopes.2 As for dCPD, a 30-residue linear sequence in its domain C mediates preS interaction.45 Transfection of dCPD cDNA into several cell lines conferred efficient DHBV binding and internalization, thus validating dCPD as a docking receptor.6 A series of studies from Dr. Schaller's lab also supported dCPD as a DHBV receptor.78910 For example, dCPD expression was reduced in DHBV infected cells, which could provide a mechanism for superinfection exclusion.10
The P protein (p120) of dGLDC as a tissue-specific binding partner for truncated DHBV L protein
Although dCPD reconstitution conferred efficient DHBV binding and internalization (as evidenced by presence of trypsin-resistant DHBV DNA), no viral DNA replication or protein synthesis could be demonstrated even in the chicken hepatoma cell line LMH, which supports DHBV DNA replication following transient transfection. Unfortunately, no duck hepatoma cell line is available. This failure suggested requirement of additional host- or tissue-specific co-factors for productive DHBV infection. In this regard, dCPD is broadly expressed in many DHBV non-infectible tissues (Table 1).12 While mapping the p170 binding site using preS deletion mutants, we identified a 120-kda duck liver protein (p120) reactive to several truncated versions of preS peptide: 1-102 (but not 1-104), 92-161, and 98-161.11 Double deletion mutants such as 80-102 (but not 80-104) and even 98-102 retained strong p120 binding, thus implicating a pentapeptide 98EAFRR102 as the p120 binding site. Interestingly, this site corresponds to a neutralizing epitope. Further site-directed mutagenesis identified residues 100, 101, and 102 as most critical for p120 interaction. In contrast to dCPD, p120 is only expressed in DHBV infectible tissues (liver, kidney, and pancreas) (Table 1).1112 Molecular cloning revealed p120 to be the p protein of duck glycine decarboxylase (dGLDC),12 which is primarily localized in mitochondria but also available on cell surface.
Distinct features of CPD and p120 as components of DHBV receptor
P120 is required for DHBV infection of primary duck hepatocytes
Several lines of evidence support the importance of p120 in DHBV lifecycle. First, DHBV infection of primary duck hepatocytes could be inhibited by preS peptide 80-104 with affinity for dCPD but not p120; it could also be inhibited by peptide 80-102 with affinity for p120 but not dCPD.11 Second, mutating residues critical for p120 binding, such as F1001V/R101L and R101I/R102D, impaired DHBV infectivity.11 Third, in vitro culture of primary duck hepatocytes resulted in rapid loss of susceptibility to DHBV infection, which correlated with declined p120 but not dCPD expression. Transfection of p120 but not dCPD cDNA to such de-differentiated hepatocytes markedly increased protein-free form of relaxed circular DNA, the precursor to cccDNA.13 Fourth, DHBV infectivity in freshly prepared duck hepatocytes could be reduced by adenovirus-mediated antisense RNA to diminish p120 expression.13 Fifth, similar effect could be achieved by a polyclonal p120 antibody added immediately following removal of the inoculum or 18 hrs later, which depleted cellular pool of p120 through cell surface association followed by internalization.13 This result also suggests that p120 acts at a post-binding step of DHBV infection.
Does DHBV entry require proteolytic cleavage of its L protein?
Sequence surrounding the p120 binding site is rich in basic residues: 95KAREAFRRYQ104. Thus, one way to create the p120 binding site is cleavage at R102 by a furin-like basic endopeptidase, which would be further destroyed by removal of R102 and R101 by a basic carboxypeptidase such as dCDP (cleavage at R97 by a different basic endopeptidase will not cause such a problem). Furin is involved in maturation of envelope protein precursors of many viruses including HIV, a prerequisite to expose the fusion peptide.14 In this regard, the Y103C/Q104F mutations immediately downstream of p120 binding site completely abolished DHBV infectivity.11 Interestingly, dCPD, a basic carboxypeptidase, has been found to transit between trans-Golgi network and cell surface.715 This is also true for furin, a basic endopeptidase.16 We found that dCPD mediated DHBV binding/endocytosis is followed by rapid viral exit, which could be minimized by co-transfection of chicken furin.17 dCPD, p120, and DHBV L protein colocalized in endosomal fraction, and endosomal enzymes could cleave the L protein on DHBV particles.17 Incubation of chicken hepatoma cell line LMH with such cleaved DHBV particles generated small amount of cccDNA. Finally, a furin inhibitor capable of blocking L protein cleavage by purified endosomal fraction could also inhibit DHBV infection of duck hepatocytes.17
HSPG serves as the low-affinity HBV receptor via its interaction with the S domain
Compared with DHBV, HBV expresses three envelope proteins: L, M (middle), and S. The M protein has an extra preS2 domain than S, while L has an extra preS1 domain than M. Both the "a" determinant (residues 124-147) in the S domain and N-terminal 75 residues in the preS1 domain are critical for HBV infectivity,1819 and corresponding antibodies can neutralize HBV infectivity. A major impediment in the search for HBV receptors has been the lack of a convenient system of in vitro infection, because primary human hepatocytes are difficult to obtain and their susceptibility to HBV infection is variable. This has been partly overcome with the establishment of an HBV infectable human hepatoma cell line (HepaRG) and demonstration of HBV susceptibility of primary Tupaia (tree shrew) hepatocytes.2021 Using these two systems, two groups independently identified heparan sulfate proteoglycans (HSPG) as the low-affinity HBV receptor.2223 First, pretreatment of HepaRG cells or primary Tupaia hepatocytes with heparinase impaired HBV infectivity. Second, heparin, the highly sulfated soluble form of heparan sulfate, could bind to HBV particles and competitively inhibit HBV infection. Third, polyethylene glycol (PEG) added at the step of inoculation could enhance HBV infectivity in HepaRG cells, and this was attributed to its augmentation of HBV binding to HSPG.22 Although the preS1 domain was initially thought to be responsible for HSPG interaction,22 further study revealed that the negatively charged HSPG interacts with two basic residues (R122 and K141) in the "a" determinant of the S domain.24 Thus, anti-S antibodies neutralize HBV infectivity by blocking virus attachment to cell surface via HSPG.
Identification of NTCP as a high-affinity functional HBV receptor interacting with the preS1 domain
To search for the high-affinity receptor(s), investigators in the field have identified more than a dozen binding partners for the preS1, preS2, or S domain of HBV envelope proteins. Unfortunately, until late 2012 none of these binding partners has been validated to confer HBV infection in an otherwise nonsusceptible cell line (for a review, see25). The hunt for HBV receptor by Dr. Wenhui Li's group from Beijing, China also began with the search for a binding partner, although a short myristoylated preS1 peptide 2-48 rather than the full-length preS1 domain was employed.26 This peptide had been previously shown by others to block HBV infection in cell culture. Their experimental design was meticulous. First, a site-specific mutant (N9K) served as a negative control. Second, the peptide was modified with an unnatural amino acid (photo-leucine) to enable zero distance cross-linking with its binding partner. Third, a two-step purification procedure for the binding partner was used to increase specificity: streptavidin for the biotin attached to the peptide, and a monoclonal antibody against part of the peptide not engaged in receptor binding. The binding partner thus identified was sodium taurocholate cotransporting polypeptide (NTCP). Silencing NTCP expression in primary Tupaia or human hepatocytes, or differentiated HepaRG cells diminished HBV infectivity. Conversely, ectopic NTCP expression rendered HepG2 cells, a human hepatoma cell line, susceptible to HBV infection.26 Therefore, NTCP was necessary and sufficient for HBV infection. What's more, NTCP silencing also impaired hepatitis D virus (HDV) infection of primary Tupaia hepatocytes and HepaRG cells, whereas NTCP transfected Huh7 cells (another human hepatoma cell line) became infectable with HDV. In this regard, HDV is a defective RNA virus employing HBV envelope proteins for virion formation and infectivity.
Further evidence supporting NTCP as a major HBV receptor in vitro
NTCP, a transmembrane protein localized on the basolateral (blood facing) side of hepatocytes, is a key transporter of bile salt from blood to the liver.27 Since the 2012 eLife paper by Yan et al.,26 a flurry of papers have been published by others which directly or indirectly support NTCP as an HBV receptor (see Table 2 for explanations).28293031323334353637383940 Also, Dr. Li's own group has extended their findings.41424344 NTCP binding to myristoylated preS1 residues 2-48 could explain why antibodies against C-terminal part of this sequence is neutralizing (the N-terminus is probably masked by myristoylation), and probably also why myristoylation is essential for infectivity (it probably partly mediates NTCP interaction by structural mimicry of cholesterol). NTCP fulfills both tissue and host specificities of HBV infection. In fact, its expression is highly dependent on hepatocyte differentiation with little expression in HepG2 or Huh7 cells. NTCP as the primary receptor could explain lack of HBV/HDV infection in crab-eating monkey and mice,2643 infection of Tupaia hepatocytes by HBV and woolly monkey hepatitis B virus,42 and ability of envelope proteins from a bat hepatitis B virus to mediate HDV infection of HepG2 cells transfected with human NTCP.28 NTCP serving as a major HBV/HDV receptor in vitro could also explain why HBV or HDV infection of HepaRG cells or primary Tupaia hepatocytes could be inhibited by high concentrations of bile salts,3041 by cyclosporine A,3334 interleukin 6,38 an antagonist of retinoic acid receptor,35 and a green tea extract.45 Other NTCP inhibitors such as ezetimibe, irbesartan, and ritonavir were also found to inhibit HDV infection in vitro.29

Evidence for or against NTCP as the major (or only) functional HBV receptor.
Are there co-factors for NTCP-mediated HBV infection and NTCP-independent pathways of viral entry?
Data presented so far clearly indicate that NTCP can serve as an HBV receptor (by reconstitution experiments), and is a major HBV receptor in vitro (by NTCP shRNAs, substrates, and inhibitors), although how HBV-NTCP interaction is activated remains to be clarified (no evidence so far that intact HBV virions or L protein can bind to NTCP). The much higher HBV infectivity in NTCP-reconstituted HepG2 in contrast to HDV preference for NTCP-Huh7 cells30 suggest additional and distinct host factors are required for NTCP-mediated HDV infection vs. HBV infection, although such co-factors may or may not work at the entry level. Since both HBV and HDV infectivity in NTCP-reconstituted cells could be further enhanced by dimethyl sulfoxide (DMSO) added to culture medium,30 some of these co-factors are expressed selectively in differentiated hepatocytes.
To establish whether there are NTCP-independent pathways of HBV entry in vitro would require complete silencing of NTCP expression at the DNA level by newly developed CRISPR-Cas system,46 rather than through shRNAs. A more critical question is whether NTCP is the major HBV receptor in vivo, considering that the three dimensional structure of the liver with hepatocytes surrounded by other cell types cannot be mimicked by a monolayer of cells cultured in plastic dishes. In fact HBV infection is quite efficient in vivo but very poor in vitro, requiring a high multiplicity of infection and addition of PEG.47 This chemical has been found to promote virus binding to HSPG22 and may trigger premature membrane fusion. Still, HBV infection in mice with humanized liver led to altered expression of genes of bile acid metabolism consistent with HBV-NTCP interaction.32 Moreover, transgenic mice expressing human NTCP could be infected with HDV, albeit in only 3% of hepatocytes.44 Thus, NTCP could serve as an HDV receptor in vivo. A genetic polymorphism in the NTCP gene, S267F (change of residue 267 from serine to phenylalanine), completely destroyed the HBV receptor function of human NTCP in cell culture.41 A molecular epidemiological study found that people without HBV infection or with resolved HBV infection (as evidence by presence of anti-HBs antibody) are more likely to have just one copy of functional NTCP gene than those remaining chronically infected,36 which supports the role of NTCP as a facilitator of HBV infection. Nevertheless, five chronic carriers of HBV had both copies of the NTCP gene mutated,36 which strongly argues for NTCP-independent HBV infection of these individuals. In this connection, it is worth mentioning that infection by cell culture-derived HBV in NTCP-reconstituted HepG2 cells is characterized by marked reduction in HBsAg secretion, and probably also virion secretion.3048 Such cells also produce atypical pattern of HBV RNAs. Whether this phenomenon is a consequence of NTCP overexpression or can be overcome by another host factor in vivo remains to be determined. To fully accept NTCP as the major HBV receptor would require a satisfactory explanation for such discordant findings.
Possible therapeutic implications
NTCP will serve as a target for therapy of chronic HBV infection if it is the primary mediator of HBV infection in vivo.48 In this regard, several NTCP inhibitors are already in clinical use for other medical conditions (hypertension, high cholesterol, retroviral infection, etc). Myristoylated preS1 peptide 2-48 under the trade name Myrcludex B could efficiently block HBV infection irrespective of the nature of HBV receptor involved. The usefulness of such entry inhibitors is dependent on the extent of hepatocyte regeneration and HBV viral load in the blood. Nucleos(t)ide analogues, by virtue of inhibiting HBV DNA replication, greatly reduces the number of infectious virions in the circulation and consequently the risk of new infection. If the block in virion secretion is complete with potent inhibitors, then adding entry inhibitors will have limited therapeutic benefit. Also, the possible side effect on cholesterol homeostasis should be monitored considering the role of NTCP in transporting bile salts. In chronic carriers most hepatocytes are already infected with HBV with cccDNA serving as the seed of continued viral protein expression and genome replication/virion secretion. Degrading cccDNA, if feasible, would have much greater therapeutic value than blocking new infection.
Acknowledgements
This work was supported by grants R21AI103648, R21AI107618, and R01AI116639 from National Institutes of Health.
Notes
Conflicts of Interest: The authors have no conflicts to disclose.
Abbreviations
cccDNA
covalently closed circular DNA
CPD
carboxypeptidase D
DHBV
duck hepatitis B virus
DMSO
dimethyl sulfoxide
GLDC
glycine decarboxylase
HBeAg
hepatitis B e antigen
HBsAg
hepatitis B surface antigen
HBV
hepatitis B virus
HCC
hepatocellular carcinoma
HDV
hepatitis delta virus
HSPG
heparan sulfate proteoglycans
L protein
large envelope protein
M protein
middle envelope protein
NTCP
sodium taurocholate cotransporting polypeptide
PEG
polyethylene glycol
S protein
small envelope protein