


INTRODUCTION
Hepatocellular carcinoma (HCC) is the sixth most prevalent cancer in the world, with approximately 0.9 million new patients reported each year. HCC is also the third leading cause of cancer-related mortality, with an estimated 830,000 deaths globally per year [1]. The incidence of HCC has continued to increase over the past few decades in many areas, including Europe, America, and Oceania [1]. HCC-related mortality remains high, at 8 per 100,000 person-years in East Asia and sub-Saharan Africa [2]. According to molecular epidemiology, many risk factors are involved in the etiology of HCC, including chronic viral hepatitis B, C, D, cirrhosis, alcohol, aflatoxin, hereditary metabolic liver disease, autoimmune hepatitis, and nonalcoholic fatty liver disease [3].
Chronic hepatitis B (CHB) is an important global health issue, affecting an estimated 350 million persons living with hepatitis B virus (HBV) infection [4]. According to the World Health OrganizationŌĆÖs global hepatitis report for 2017, the estimated prevalence of chronic HBV infection in the whole population was 6.2% in the Western Pacific, 6.1% in sub-Saharan Africa, 3.3% in the Eastern Mediterranean, and 2.0% in Southeast Asia [5]. Although vaccination campaigns have reduced the rate of new infections in infants and children [6], chronic HBV infection remains hyperendemic in adults in some areas. For example, the prevalence of the hepatitis B surface antigen (HBsAg) in children decreased from 7.4% to 0.4% after the introduction of universal HBV vaccines in Taiwan [7]. However, the prevalence of HBsAg remains at 13.7% in adults in Taiwan [8]. Long-term hepatic inflammation of CHB leads to the development sequelae, including end-stage liver disease and HCC [9,10]. Because of geographic differences in the prevalence of CHB, the contribution of HBV to HCC development has regional variations. In a systematic review, HBV was found to be responsible for more than 50% of HCC cases in most Asian countries and Africa, including Taiwan, China, Korea, Thailand and Egypt. The proportion of HBV-related HCC was highest in Greece at 56%, but less than 20% in other European countries [11].
Over the past two decades, the development of antiviral therapies that delay or inhibit the progression of hepatic necroinflammation has further reduced the incidence of HCC and improved the long-term outcomes of CHB patients receiving antiviral treatment [12]. However, while antiviral therapy can reduce the risk of HBV-related HCC, it has not eradicated the disease. In this review, the risk of HCC in untreated and treated patients with CHB will be summarized and discussed.
RISK FACTORS ASSOCIATED WITH HBV-RELATED HEPATOCARCINOGENESIS
Role of hepatitis B viral factors in hepatocarcinogenesis
Hepatitis B viral load, host factors, and environmental factors are all involved in HBV-related hepatocarcinogenesis (Table 1) [3,13,14]. Among the viral factors, hepatitis B viral load has been confirmed to be the driving force for the risk of HCC development in the natural course of CHB [15-19]. Long-term immune responses in hosts against HBV cause chronic hepatic inflammation and lead to a continuous cycle of hepatocyte death and regeneration. Subsequently, the risk of cirrhosis and HCC will be substantially increased [20]. However, hepatitis B viral load thresholds and the risk of HCC need to be clarified. In the Risk Evaluation of Viral Load Elevation and Associated Liver Disease/Cancer-Hepatitis B Virus (REVEAL-HBV) study, 85% of non-cirrhotic patients tested negative for the hepatitis B viral protein HBeAg and had HBV DNA levels below 5 log10 IU/mL. There is a dose-response relationship between HBV DNA concentration and HCC [15]. In a Korean cohort of 6,949 non-cirrhotic patients, patients with moderate levels of HBV DNA (6ŌĆō7 log10 IU/mL in HBeAg-positive patients and >5 log10 IU/mL in HBeAg-negative patients) had the highest HCC risks compared with those with low (Ōēż4 log10 IU/mL) and high (>8 log10 IU/mL) levels of HBV DNA. Patients with moderate replicative activity (HBV DNA 4.0ŌĆō8.0 log10 IU/mL) had the highest risk of HCC, irrespective HBeAg status [21]. This phenomenon can be explained by the emergence and expansion of a clonal premalignant hepatocyte [22-25]; the observation that a moderate hepatitis viral load is associated with significant inflammation [26]ŌĆömost CHB patients with a high viral load eventually improve to a moderate viral load after a flare-up of hepatitisŌĆöand subsequently an increased risk of HCC; and the integration of HBV DNA into human chromosomes [24,27].
In the life cycle of HBV replication, HBV DNA can integrate into host genomic DNA, promote genomic instability, and induce insertional mutagenesis of HCC-related genes [28]. HBV integration, which can occur at every phase of the natural course of HBV infection, is detected in more than 75% of CHB patients with HCC [29,30]. Li et al. [31] investigated the junctional fragments at the integration site, defined as virus-host chimeric DNA, in HBV-related HCC. They found that the common location of integration included the TERT, CCNE1, and MLL4 genes. In addition, circulating tumor-specific, virus-host chimera DNA has been detected in 97.7% of CHB patients with HCC [31]. Recently, P├®neau et al. [32] investigated the frequency of HBV integration and host genome rearrangement in pairs of HCC and non-HCC liver tissue. The results revealed that the insertion of HBV into human genes triggers two distinct oncogenic mechanisms. First, HBV-embedded viral enhancers near cancer-driver genes can lead to overexpression of these genes. Second, frequent human genomic rearrangements near HBV integration sites can lead to sequence alterations in cancer-driver genes [32]. HBV integration may therefore predispose a patient to the possibility of host genetic alterations responsible for HCC carcinogenesis [33].
HBV-encoded proteins have a direct oncogenic potential. Among them, the HBV X protein is the most potent. Through a multifunctional regulatory mechanism, including interactions with signal-transduction pathways, epigenetic modification of DNA repair gene, regulation of mitochondrial pathways of apoptosis, and activation of non-coding RNAs and microRNA, the HBV X protein plays a role in hepatocellular carcinogenesis [34].
Several HBV mutations occurring naturally during virus evolution have also been shown to be associated with HCC development [35]. In large-scale, long-term, follow-up cohorts from hospitals and community, basal core-promoter double mutations were confirmed as risk factors for development of cirrhosis and HCC [36,37]. HBV pre-S1 and preS2 deletion mutations induce HBV surface protein synthesis dysregulation and may activate endoplasmic reticulum stress. Increasing endoplasmic reticulum stress can cause hepatocyte inflammation, cell damage, and fibrosis, and therefore may contribute to HCC formation [38]. A recent retrospective case-control study in Taiwan revealed that preS mutations were significantly associated with HCC risk (hazard ratio [HR], 3.210; 95% confidence interval [CI], 1.072ŌĆō9.613; P=0.037), even in patients with low serum levels of HBV DNA or alanine aminotransferase (ALT) (HR, 2.790; 95% CI, 1.133ŌĆō6.873; P=0.026) [39].
HBV evolution and the diversity of the genomic sequence has led to a variety of genotypes, of which 10 well-known genotypes (AŌĆōJ) have been defined [35]. HBV genotypes B and C are predominant in Asian countries, whereas genotypes A and D are common in Europe. Genotype E is restricted to the western part of sub-Saharan Africa and genotype F is prevalent in Latin America [40]. Untreated patients with genotype C infection have higher hepatitis B viral loads, hepatic inflammation severity, and increased risks of HCC occurrence compared with those with genotype B infection. Patients infected with HBV genotypes D and F have a higher risk of HCC development than those with genotype A infection [35]. Recently, a 35-year follow-up study of a Native Alaskan population revealed that the HBV genotype had a strong correlation with the occurrence of HCC. The risk of HCC was significantly higher in genotypes F, C, and A compared with genotypes B and D [41]. Accumulating evidence indicates that the HBV genotype is an important risk factor in risk stratification of HBV-related HCC.
Role of host factors in hepatocarcinogenesis
Clinical epidemiological studies have demonstrated that several host factors are associated with HBV-related HCC, including gender, age, alcohol consumption, smoking, type 2 diabetes mellitus, and concomitant hepatic steatosis. Male dominance is a special feature of HBV-related HCC. The ratio of males to females is approximately 2.9:1 [42]. Sex hormones and the active androgen pathway are associated with HCC development in male HBV carriers. The mechanisms of sex hormones in hepatocarcinogenesis regulate the hostŌĆÖs immune response to HBV, influence the expression of HBV-targeting microRNAs, and a play a role in HBV chromosomal integration [43]. A meta-analysis of 66 studies with more than 340,000 patients revealed that male gender (relative risk [RR], 2.7; 95% CI, 2.1ŌĆō3.3), increasing age (RR, 1.7; 95% CI, 1.4ŌĆō2.1, for a 10-year increase), and alcohol consumption (RR, 2.1; 95% CI, 1.0ŌĆō4.6) significantly increased the incidence of HCC in patients with CHB [44]. In addition, several large-population studies have found that non-alcoholic fatty liver disease (NAFLD) and type 2 diabetes mellitus are associated with an increased in HCC incidence and mortality in patients with CHB. For example, a long-term follow-up study in Taiwan demonstrated that HBV carriers with more than 3 metabolic risk factors (obesity, diabetes, hypertriglyceridemia, and high blood pressure) had significantly higher 10-year cumulative incidence rates of HCC compared with those with 2 or fewer metabolic risk factors (13.6% vs. 4.83%; adjusted HR, 2.32; 95% CI, 1.18ŌĆō4.54). The 10-year cumulative incidence of HCC was particularly high in HBV carriers who smoke and have more than 3 metabolic risk factors compared with smokers with no metabolic risk factors (25% vs. 3.87%, P<0.0001) [45]. A retrospective study of 270 CHB patients found that the risk of developing HCC increases by a factor of 7.3 in those with concomitant, biopsy-proven NAFLD [46]. A nationwide, long-term, follow-up cohort study in Korea also revealed that concomitant NAFLD significantly increased the risk of HCC in patients with chronic hepatitis B and C [47].
Host genetic alteration also plays an important role in hepatocarcinogenesis. Several genes with somatic mutations, including telomerase reverse transcriptase, p53, and ╬▓-catenin are associated with hepatocarcinogenesis [3,48]. For example, the frequency of telomerase reverse transcriptase promoter mutations increased from 6% to 19% in hepatic dysplastic nodules to 42% to 61% in HCC [49]. Several single nucleotide polymorphisms (SNPs) are associated with an increased risk of HCC through alteration of molecular signaling pathways of hepatocarcinogenesis [48]. As an example, an SNP in STAT4 gene is associated with a risk of HCC in patients with CHB [50].
HBV-RELATED HCC IN UNTREATED PATIENTS
HCC Incidence in untreated patients
In the natural course of CHB, approximately 2ŌĆō10% of patients progress to cirrhosis annually. Although the annual incidence of HCC in non-cirrhotic patients is less than 1%, the annual incidence of HCC increases to 3% in cirrhotic patients. Without proper treatment, 25ŌĆō40% of CHB patients face the threat of an early death [14]. In a meta-analysis of 66 studies involving more than 340,000 untreated patients, the estimated HCC incidence rates per 100 person-years were 0.03ŌĆō0.17 in inactive carriers (defined as normal serum ALT and HBeAgnegative/anti-HBe-positive), 0.07ŌĆō0.42 in asymptomatic carriers (defined as no symptoms or signs of CHB), 0.12ŌĆō0.49 in chronic hepatitis (defined as the presence of histology or clinical features of chronic inflammation), and 2.03ŌĆō3.37 in cirrhosis [44]. Although the incidence of HCC was much higher in patients with cirrhosis, patients without cirrhosis remain at considerable risk. In a long-term follow-up study of 3,366 untreated non-cirrhotic CHB patients, 39% had elevated ALT levels but a low hepatitis B viral load, or a high viral load but normal ALT levels at baseline (termed an indeterminate phase). Compared with patients with persistently low HBV DNA and normal ALT levels (a so-called inactive phase), those in the indeterminate phase had a significantly higher HCC incidence (10-year cumulative rate of 4.6% vs. 0.5%; P<0.0001). The HR of HCC increased up to 14-fold [51]. An early longitudinal large-cohort study in Taiwan followed 1,932 inactive carriers and 18,137 HBsAg-negative controls. After a mean follow-up period of 13 years, the annual incidence of HCC was 0.06% in inactive carriers compared with 0.02% in a control group. The relative risk of HCC was 4.6 for inactive HBV carriers compared with non-infected controls [52].
Biomarkers of HBV-related HCC for untreated patients
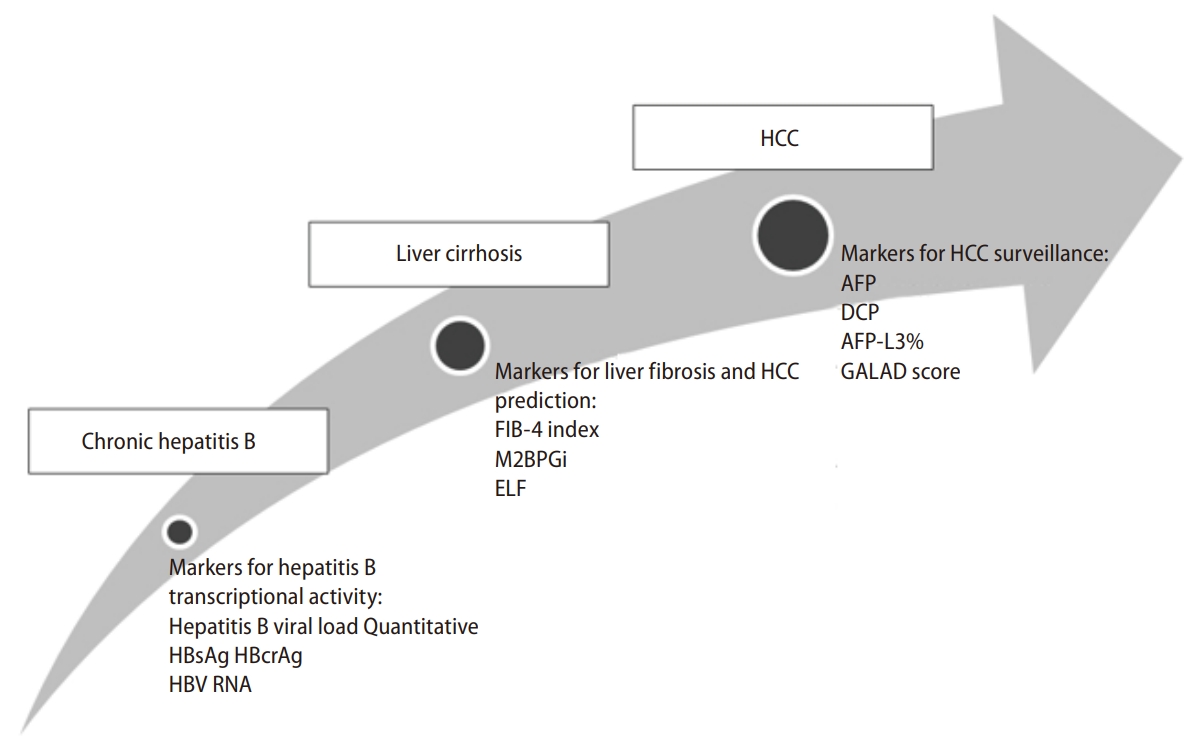
Due to the poor prognosis of HCC, which has an overall 5-year survival rate of 20% [53], serum markers for the diagnosis and prediction of HCC are urgently needed. The serum markers can be divided into 3 categories: those for hepatitis B transcriptional activity, those for liver fibrosis and HCC prediction, and those for HCC surveillance (Fig. 1).
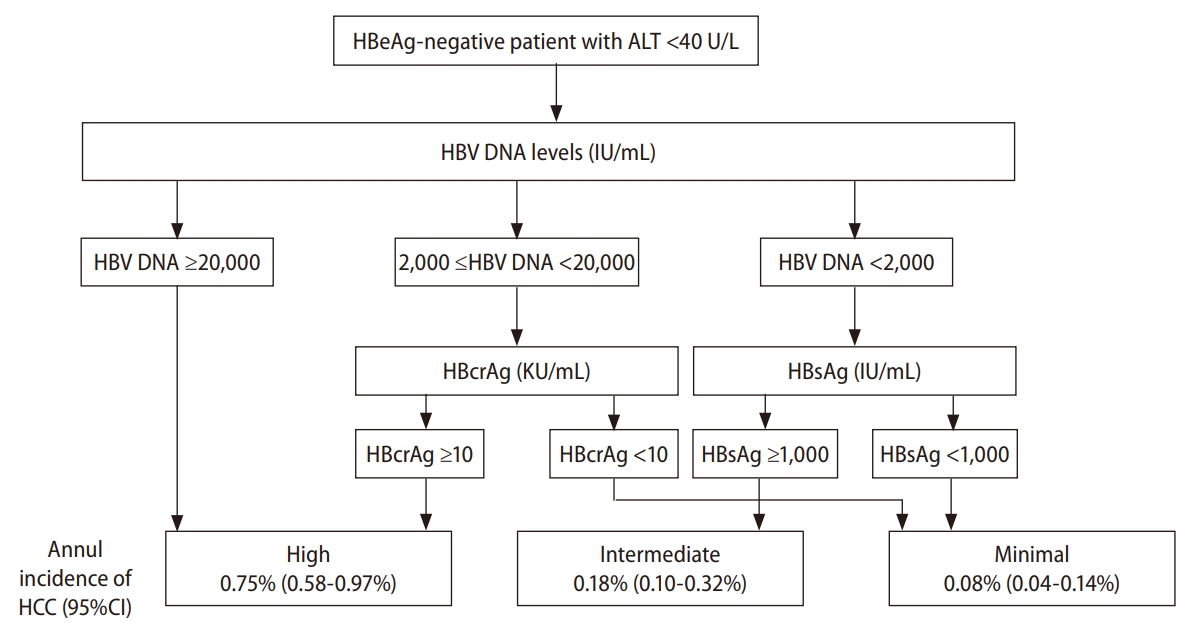
The first category comprises biomarkers for the assessment of hepatitis B transcriptional activity, including hepatitis B viral load, quantitative HBsAg, hepatitis B core-related antigen (HBcrAg), and HBV RNA. Hepatitis B viral load is the strongest predictor of HCC. The dose-effect relationship of HBV DNA concentration and risk of HCC was initially reported in the REVEAL-HBV study. Over a follow-up period of 11 years, the cumulative incidence rates of HCC increased from 1.3% for patients with HBV DNA of less than 60 IU/mL to 14.9% for those with more than 200,000 IU/mL. The cut-off value at which the risk of HCC began to increase was 2,000 IU for HBeAg-negative, non-cirrhotic patients [15]. However, the predictive value of HBV DNA is less powerful in patients with cirrhosis. In a Korean cohort, the 5-year cumulative incidence rate of HCC was still 9.6% in compensated cirrhotic patients with low hepatitis B viremia [54]. Fortunately, quantitative HBsAg is associated with a complementary effect to HBV DNA levels for predicting HCC risks in patients with a low viral load. In a long-term follow-up study of 2,688 non-cirrhotic patients with persistent HBV DNA levels <2,000 IU/mL, patients with an HBsAg level Ōēź1,000 IU/mL had a much higher risk of HCC development (HR, 13.7) compared with those with an HBsAg level <1,000 IU/mL [55]. These findings were further confirmed by the REVEAL studyŌĆÖs HBV cohort. Patients with both HBsAg <1,000 IU/mL and HBV DNA <2,000 IU/mL had lower HCC risks, with an HR of 0.36, compared with those with either HBsAg Ōēź1,000 IU/mL or HBV DNA Ōēź2,000 IU/mL [56].
HBcrAg is a newly discovered hepatitis B viral biomarker, which contains the hepatitis B core antigen, HBeAg, and a precore protein (p22cr) [57]. Although HBcrAg has been known for 20 years, it has only been used recently in clinical practice to monitor HBV disease progression and response to antiviral therapy [58,59]. Tada et al. [60] reported that HBcrAg was significantly associated with HCC occurrence in untreated patients. In a long-term follow-up study, Tseng et al. [61] determined that HBcrAg levels stratified the HCC risk in HBeAg-negative, non-cirrhotic CHB patients with intermediate viral load (2,000ŌĆō19,999 IU/mL). Patients with HBcrAg >10 IU/mL had a higher risk of HCC incidence (HR, 6.29) compared with those with low level of HBcrAg [61]. Tseng et al. [62] further investigated the association of HBcrAg and HCC risk in HBeAg-negative, non-cirrhotic carriers at the indeterminate clinical phase. The cumulative HCC incidence was much higher in patients with high HBcrAg levels (>10,000 U/mL) compared with those with low HBcrAg levels (<10,000 IU/mL) (5.33% vs. 0.51%) [62]. Accordingly, the combination of serum biomarkers, including HBV viral load, quantitative HBsAg, and HBcrAg, could be used for HCC risk stratification in HBeAg-negative CHB patients with a low serum level of ALT (Fig. 2).
HBV RNA was positively correlated with intrahepatic cccDNA [63]. HBV RNA was correlated with HBV DNA, HBsAg levels, HBeAg status, serum ALT, and HBV genotype [64]. Serum HBV RNA levels were also associated with virological responses to antiviral therapy [65,66]. HBV RNA can be used to distinguish inactive from active hepatitis in CHB patients. In HBV-related HCC, tumor tissues with detectable HBV RNA had less tumorous microvascular invasion and better prognosis [67]. However, the application of HBV RNA to the management of HBV-related HCC requires further investigation.
The second category comprises biomarkers for liver fibrosis and HCC prediction. Hepatic necroinflammation caused by chronic HBV infection is the major cause of hepatic fibrogenesis and carcinogenesis. Several host inflammatory biomarkers have been developed to evaluate fibrosis severity and can predict HCC risks. Among them, the fibrosis-4 (FIB-4) index, which is measured by age, aspartate aminotransferase and ALT levels, and platelet count, is correlated with histologic fibrosis severity in patients with CHB [68]. In a long-term follow-up of 2,075 untreated patients with CHB, the FIB-4 index was positively correlated with the incidence of HCC. Patients with a FIB-4 index >1.29, had a higher risk of HCC compared with patients with a FIB-4 index <1.29 (HR, 5.56; 95% CI, 3.93ŌĆō7.86) [69].
The Mac-2 binding protein glycosylation isomer (M2BPGi), a glyco-biomarker, is associated with the severity of liver fibrosis [70]. Furthermore, M2BPGi is a powerful predictor of HCC. In a retrospective study of 1,070 untreated CHB patients, Liu et al. [71] found that M2BPGi levels were an independent risk factor in HCC development. Patients with an M2BPGi cut-off index Ōēź2 had a significantly higher risk of HCC occurrence within 5 years of follow-up [71]. Similarly, Kim et al. [72] reported that the M2BPGi level was associated with an HCC risk in untreated patients with CHB. In a study of 207 CHB patients after spontaneous HBeAg seroconversion, Mak et al. [73] found that patients with persistently high M2BPGi levels (cut-off index >0.68) at 3, 5, and 10 years after HBeAg seroconversion had a significantly higher incidence rate of HCC compared with those with persistently low M2BPGi levels. The HR of HCC with a high M2BPGi was 4.66 (P=0.018). Taking 0.68 as the cut-off value had a high accuracy for predicting HCC, with an area under the receiver operating characteristic (AUROC) of 0.883 [73]. Taken together, the M2BPGi has a high specificity for predicting HCC in untreated CHB patients.
The enhanced liver fibrosis (ELF) score, consisting of age, hyaluronic acid, amino-terminal propeptide of type III collagen, and tissue inhibitor of metalloproteinase 1, has a high diagnostic specificity for identifying advanced fibrosis and cirrhosis [74,75]. In a follow-up study of 170 CHB patients, patients with a high ELF score had a higher risk of liver-related complications (hepatic decompensation, HCC, and liver-related death) compared with those with low ELF score. The predictive value of ELF scores for HBV-related liver complications by AUROC was 0.808 [76].
The third category comprises biomarkers for surveillance of HCC, including ╬▒-fetoprotein (AFP), des-gamma carboxy-prothrombin (DCP) and lectin-bound AFP (AFP-L3%). Serum AFP has been used clinically for HCC surveillance for more than 50 years. In a systematic review of 5 studies, AFP at a cut-off value of 20 ng/mL had high specificity (80ŌĆō94%) for HCC diagnosis [77]. DCP is a valuable biomarker and can be used as a complement of AFP for HCC diagnosis. In several case-control studies, DCP exhibited high sensitivity and specificity for HCC detection [78-80]. The AUROC value of combining AFP and DCP for HCC diagnosis was high, at 0.90 (95% CI, 0.87ŌĆō0.94) [80]. In a meta-analysis of 12 studies, AFP-L3% demonstrated a specificity of 0.929 (95% CI, 0.916ŌĆō0.940) and a sensitivity of 0.483 (95% CI, 0.459ŌĆō0.507) for diagnosis of HCC [81]. In a large-scale, case-control study, the AUROC values of AFP, DCP, and AFP-L3 for the diagnosis of early-stage HCC were 0.80 (95% CI, 0.77ŌĆō0.84), 0.72 (95% CI, 0.68ŌĆō0.77), and 0.66 (95% CI, 0.62ŌĆō0.70), respectively [82]. Because of the limitation presented by a single biomarker in HCC surveillance, the GALAD score, a serum biomarkerŌĆōbased model, was developed to improve the early diagnosis of HCC. The components of the GALAD score include gender, age, AFP, DCP, and AFP-L3 [83]. In a phase 2 cohort study, the AUROC of the GALAD score for diagnosis of HCC was 0.95 (95% CI, 0.93ŌĆō0.97) [84]. Recently, Singal et al. [85] demonstrated that a longitudinal GALAD score was superior to a single time-point GALAD score in HCC diagnosis (c-statistic, 0.85 vs. 0.79).
HBV-related HCC risk scores for untreated patients
Since the identification of risk factors for HBV-related HCC, several prediction models for HBV-related HCC risk stratification in untreated patients have been developed (Table 2) [18,19,86-92]. The majority of prediction models consist of host factors and hepatitis B viral factors. For example, the risk estimation for hepatocellular carcinoma in chronic hepatitis B (REACH-B) risk score includes age, gender, ALT level, HBeAg status, and HBV DNA as components. A 17-point score was used to assess HCC risk at 3, 5, and 10 years. The predictive accuracies for HCC risk at 3, 5, 10 years by AUROC were 0.811, 0.796, and 0.769, respectively [86]. Assuming liver fibrosis is an important risk factor for HCC development, several prediction models also include liver stiffness measurement (LSM) using noninvasive methods. The LSM HCC risk score incorporates age, albumin level, HBV DNA, and liver stiffness values using transient elastography. The AUROC for predicting HCC risk at 5 years was 0.83 [89]. Clinically, high-risk HBV carriers for HCC development can be identified by HCC prediction models, which can reduce the burden of patients in need of HCC surveillance [93].
Most HCC prediction models were established using data from Asian patients, and whether they can be applied to Western populations has yet to be determined. In a cohort of CHB patients of different ethnicities, the guide with age and gender (GAG)-HCC, Chinese University (CU)-HCC and REACH-B scores had similar predictive ability for HCC in Asian and non-Asian populations [94]. A large-scale study is warranted to determine the effect of ethnic differences in HCC prediction models.
HBV-RELATED HCC IN TREATED PATIENTS
HCC incidence in treated patients
Although eradication of HBV remains a challenge, antiviral therapies can induce hepatic fibrosis regression and reduce the incidence of HCC. According to international guidelines, current antiviral therapies include interferon-╬▒ (standard or pegylated form) and nucleos(t)ide analogs (NAs) [95-97]. Several meta-analyses have demonstrated that responders with interferon-╬▒ therapy decrease the risk of HCC developing by 34ŌĆō41% compared with untreated patients, particularly in Asian populations [98-100]. Studies from Taiwan and China show that interferon-╬▒ monotherapy or sequential therapy was more effective in reducing HCC incidence than NA monotherapy. HCC prevention was more obvious in high-risk patients [101,102].
Due to the convenience of oral administration, fewer side effects, and profound suppression of HBV replication, NAs have been widely used to treat patients with chronic HBV infection. Several population- and hospital-based cohorts of Asian populations indicate that entecavir therapy significantly reduced HCC incidence in CHB patients by 50ŌĆō60% [12]. Similarly, patients receiving long-term tenofovir disoproxil fumarate had a reduced risk of HCC (HR, 0.23ŌĆō0.46) compared with untreated patients [103-105]. Another hospital-based cohort revealed that the annual HCC incidence was comparable for patients treating with tenofovir disoproxil fumarate and tenofovir alafenamide [106]. In a recent meta-analysis consist of 31 studies and 119,053 CHB patients, no significant difference in HCC incidence was found among patients receiving entecavir or tenofovir disoproxil fumarate [107]. However, several studies have shown that patients given tenofovir disoproxil fumarate had a lower rate of HCC development than those given entecavir [108,109]. For example, a meta-analysis of 14 studies and 263,947 person-years of follow-up revealed that patients receiving entecavir had a higher HCC risk compared with those receiving tenofovir disoproxil fumarate treatment (HR, 1.27; 95% CI, 1.01ŌĆō1.60, P=0.04) [108]. Whether different antivirals lead to distinct risks of HCC developments remains to be seen.
Although antiviral therapy achieves long-term viral suppression, HCC still presents, even in patients with undetectable HBV DNA. In a retrospective study of 875 CHB patients receiving long-term NA treatment, the 5-year cumulative HCC incidence rate was 7.5% for patients with maintained undetectable HBV DNA and 14.3% for patients with a low viral load (<2,000 IU/mL) [110]. A recent study conducted by Burdette et al. [111] found that few patients had no quantifiable, but detectable HBV DNA after receiving long-term NA treatment. In addition, these unquantifiable HBV DNA samples were still infectious in a mouse model [111]. Because current NA options cannot eliminate cccDNA, treated patients with residual low levels of HBV DNA or intrahepatic cccDNA are still at risk of liver disease progression to cirrhosis and HCC.
Biomarkers of HBV-related HCC for treated patients
Due to long-term viral suppression, hepatitis B viral loads cannot be used as a marker of HBV transcriptional activity in patients undergoing NA therapy. In a previous study with treatment-naïve CHB patients, Testoni et al. [112] found that serum levels of HBcrAg were correlated with hepatitis B viral loads and intrahepatic cccDNA levels. In a study of CHB patients with liver biopsy at baseline and at 96 weeks after NA therapy, Wang et al. [113] reported that the serum levels of HBcrAg and intrahepatic cccDNA concentrations were positively correlated even when HBV DNA levels were low or undetectable after NA treatment. HBcrAg may therefore reflect the transcriptional activity of intrahepatic cccDNA, and could serve as a biomarker for evaluation of CHB progression. In a case-controlled study, NA-treated CHB patients with subsequent HCC occurrence had significantly higher post-NA-treatment HBcrAg levels compared with patients who did not develop HCC. The HR of HCC was up to 3.27 [114]. In a long-term follow-up study of 1,268 CHB patients receiving NA therapy, those with a high level of HBcrAg after 1 year of NA treatment were associated with HCC development, regardless of HBeAg status [115].
M2BPGi has been used to predict HCC in CHB patients receiving antiviral therapy. In a long-term follow-up study of 899 CHB patients receiving NA therapy, Tseng et al. [116] reported that the pre-treated serum M2BPGi level was a risk factor of HCC development during a mean follow-up of 7 years. HCC risks increased by quartiles of M2BPGi. At a cut-off index value of 1.73, the HR of HCC development in patients with a high baseline level of M2BPGi was 5.8 (95% CI, 3.5ŌĆō9.6) [116]. In a separate study of 147 CHB patients under NA therapy, Murata et al. found that the serum level of M2BPGi decreased during the treatment period. Patients with high M2BPGi levels (cut-off index >1.5) at 48 weeks of NA therapy had a higher risk of HCC occurrence. The 5-year cumulative incidence rates of HCC in patients with low and high on-treatment M2BPGi levels were 4.2% and 25.6%, respectively (P<0.001) [117]. M2BPGi can stratify the HCC risk even in high-risk groups of HCC, such as patients with cirrhosis. Su et al. [118] included NA-treated patients with compensated cirrhosis; those with an on-treatment M2BPGi cut-off index >3 at the time point of virologic remission were at a significantly higher risk of HCC development compared with those with a low level of M2BPGi (HR,1.58; 95% CI, 1.19ŌĆō2.10, P=0.002) [118]. These studies indicated that both baseline and on-treated M2BPGi are valuable biomarkers for HCC risk prediction in NA-treated CHB patients.
The FIB-4 index, calculated from a combination of routine laboratory tests, is a widely used non-invasive fibrosis biomarker. In a retrospective study of 1,936 non-cirrhotic patients with long-term NA therapy, Tseng et al. [119] found that the on-treatment FIB-4 index at 1 year after NA therapy was a predictor of HCC development. Patients with an on-treatment FIB-4 index >1.3 had a significantly higher risk of HCC development compared with those with an FIB-4 <1.3 (HR, 4.87; 95% CI, 2.48ŌĆō9.55) [119]. Similarly, Wang et al. [120] conducted a retrospective study of CHB patients receiving long-term NA therapy. Cirrhotic patients with an FIB-4 index >2.28 at 1 year after NA therapy and an FIB-4 index increase after NA therapy compared with before treatment had the highest 5-year cumulative HCC incidence (35.3%) [120]. Taken together, these findings support the use of on-treatment FIB-4 index as a biomarker for HCC risk stratification in NA-treated patients.
HBV-related HCC risk scores for treated patients
As previously mentioned, baseline HBV DNA levels are parabolically associated with a risk of HCC development. Patients with a moderate viral load (4ŌĆō8 log10 IU/mL) had the highest risk of HCC occurrence [21]. Recently, Choi et al. [121] conducted a Korean cohort study containing HBeAg-positive, non-cirrhotic patients with HBV DNA levels >5 log10 IU/mL receiving NA treatment. During a median follow-up of 5.7 years, the on-treatment HCC risk was inversely associated with baseline hepatitis B viral load [121]. The impact of baseline levels of HBV DNA on HCC risk in treated patients is therefore unclear, and further validation of this study and a new HCC risk prediction model, including pretreatment baseline HBV DNA levels based on these results, may be required. Currently, several HCC prediction models that do not contain pretreatment hepatitis B viral loads have been developed for CHB patients under NA treatment (Table 3) [122-128]. For example, the PAGE-B model enrolled 1,851 Caucasian CHB patients who received NA treatment for more than 1 year. This predictive model was composed of age, gender, and platelets. The risk of 5-year cumulative HCC incidence rate was stratified into 3 grades: 0% for low-risk patients (PAGE-B score Ōēż9), 3ŌĆō4% for moderate-risk patients (PAGE-B score 10ŌĆō17), and 16ŌĆō17% for high-risk patients (PAGE-B score Ōēź18) [122]. Furthermore, the PAGE-B model was validated in a cohort of patients representing various ethnicities. The AUROC of the PAGE-B model for HCC prediction reached 0.91 among the ethnically mixed population [129]. Kim et al. [123] also added serum albumin levels to the variables of PAGE-B model for Asian CHB patients and named it the modified PAGE-B (mPAGE-B) model. Among 2001 CHB patients, including 20% with cirrhosis, the predicted 5-year cumulative incidence rates of HCC were 0.7%, 5.1%, and 18.4% in the patients at low risk (mPAGE-B Ōēż8), intermediate risk (mPAGE-B 9ŌĆō12), and high risk (mPAGE-B Ōēź13), respectively. According to the AUROC curve, the accuracy of mPAGE-B predictions of HCC risk at 5 years was 0.82 [123].
Other risk factors, including alcohol consumption and comorbidities such as diabetes mellitus also contribute to HCC development in CHB patients [130]. In the REAL-B model by Yang et al. [128], 7 risks factors were identified in 8,048 Asian CHB patients undergoing NA therapy. The risk variables included gender, age, alcohol use, diabetes, baseline cirrhosis, platelet count, and serum level of AFP. The range of REAL-B score was 0ŌĆō13 points. According to the 3-year predicted risk of HCC, a REAL-B score of 0-3 points was classified as a low risk (3-year HCC risk <1%), 4ŌĆō7 points as a moderate risk (3-year HCC risk 1ŌĆō5%), and 8ŌĆō13 points as a high risk (3-year HCC risk >5%). The 3-year predicted risk of HCC was 0.14% and 65.6% for those with the lowest score of 0 and the highest score of 13, respectively. The accuracy of HCC risk prediction at 3, 5, and 10 years by AUROC curves was greater than 0.8 [128].
Liver fibrosis regression after long-term antiviral therapy was confirmed by liver biopsy in several studies [131-133]. The on-treatment improvement of liver fibrosis can be assessed via LSM by ultrasound-based elastography. Chon et al. [134] found that liver stiffness values fell significantly over time in CHB patients receiving long-term antiviral therapy. Furthermore, the liver stiffness-based HCC prediction models using baseline and on-treatment liver stiffness both achieved significantly predictive abilities for HCC development [135]. For example, in an earlier Korean study of 192 CHB patients who achieved complete virologic response after antiviral therapy, the liver stiffness value at the time of complete virologic response was used to replace HBV DNA and incorporated into the REACH-B predictive model, which was referred to as the modified REACH-B scoring model. The predictive effect of the modified REACH-B scoring model on HCC risk at a follow-up of 3 years was better than that of the REACH-B model (AUROC value, 0.814 vs. 0.629) [136]. Similarly, liver stiffness values were incorporated into the PAGE-B prediction model of Korean CHB patients receiving anti-viral therapy and named the modified PAGELS-B model. The performance of the modified PAGELS-B model for predicting HBV-related HCC risk was superior to those of the PAGE-B and mPAGE-B models (AUROC value 0.760 vs. 0.714 and 0.716, respectively) [137]. Dynamic assessment of HCC risks through liver stiffnessŌĆōbased prediction models should therefore be incorporated into HCC surveillance of patients receiving antiviral therapies.
Based on HCC risk-predicting models, a personalized HCC surveillance plan should be provided to CHB patients receiving antiviral therapy. For high-risk patients, in addition to close follow-ups, continuing antiviral therapy will be necessary.
PERSPECTIVES AND CONCLUSIONS
Since the discovery of the HBsAg in 1965, tremendous efforts have been made to control the burden of CHB. Global HBV vaccination programs have significantly decreased the neonatal HBV infection rate and the incidence of primary liver cancer in children [138]. For example, the annual incidence rate of primary liver cancer decreased to zero since 2011 in Taiwan, the first country to implement a hepatitis B vaccination program [139]. For patients with active CHB, long-term NA therapy has markedly decreased the risk of HCC development. However, several challenges remain before HBV-related HCC can be eliminated. First, the benefit of antiviral treatment in HCC reduction remains unclear among patients with normal or minimally higher serum levels of ALT. In a randomized trial conducted by Hsu et al. [140], 160 treatment-na├»ve, non-cirrhotic patients with serum ALT levels between 1- and 2-fold the upper limit of normal were randomized to receive tenofovir disoproxil fumarate or placebo treatment for 3 years. Tenofovir disoproxil fumarate significantly reduced the risk of fibrosis progression (RR, 0.56; 95% CI, 0.35ŌĆō0.88; P=0.013) [140]. A multinational, multicenter, open-label, phase 4 trial (ATTENTION study; NCT03753074) is currently underway to clarify whether antiviral therapy decreases the risk of HCC development in this special clinical setting. Second, NAFLD or metabolic dysfunctionŌĆōassociated fatty liver disease (MAFLD) are associated with an increased risk of endŌĆÉstage liver disease and HCC. The high prevalence of NAFLD/MAFLD has resulted in the increased coexistence of NAFLD/MAFLD with CHB in area with HBV endemic. In a community-based cohort study, Yu et al. [45] investigated the HCC risk in 1,690 CHB patients with different metabolic risk factors. Compared with patients with 1ŌĆō2 metabolic risk factors, patients with more than 3 risk factors had a significantly higher risk of developing HCC, with an adjusted HR of 2.32 (95% CI, 1.18ŌĆō4.54) [45]. The impact of NAFLD/MAFLD on the risk of HCC is increasingly important in CHB patients, particularly those with undetectable HBV DNA after antiviral therapy.
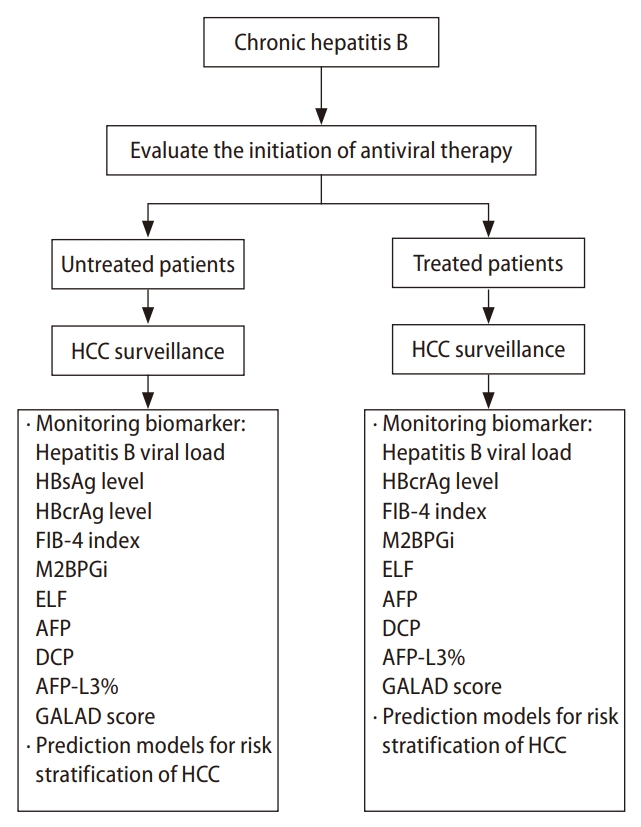
In the management of CHB, it is essential to evaluate the severity of hepatic inflammation and liver fibrosis and to provide timely antiviral treatment for the reversion of disease progression and reduction of HCC risk [141]. Monitoring existing and emerging biomarkers for risk of HBV-related HCC and applying prediction models for HCC risk stratification will provide personalized HCC surveillance for untreated and treated CHB patients (Fig. 3). Application of these strategies of prevention, early diagnosis and curative treatment of HCC should help reduce the threat of HCC and prolong the survival of CHB patients.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





