Toward a complete cure for chronic hepatitis B: Novel therapeutic targets for hepatitis B virus
Article information

Abstract
Hepatitis B virus (HBV) affects approximately 250 million patients worldwide, resulting in the progression to cirrhosis and hepatocellular carcinoma, which are serious public health problems. Although universal vaccination programs exist, they are only prophylactic and not curative. In the HBV life cycle, HBV forms covalently closed circular DNA (cccDNA), which is the viral minichromosome, in the nuclei of human hepatocytes and makes it difficult to achieve a complete cure with the current nucleos(t)ide analogs and interferon therapies. Current antiviral therapies rarely eliminate cccDNA; therefore, lifelong antiviral treatment is necessary. Recent trials for antiviral treatment of chronic hepatitis B have been focused on establishing a functional cure, defined by either the loss of hepatitis B surface antigen, undetectable serum HBV DNA levels, and/or seroconversion to hepatitis B surface antibody. Novel therapeutic targets and molecules are in the pipeline for early clinical trials aiming to cure HBV infection. The ideal strategy for achieving a long-lasting functional or complete cure might be using combination therapies targeting different steps of the HBV life cycle and immunomodulators. This review summarizes the current knowledge about novel treatments and combination treatments for a complete HBV cure.
INTRODUCTION
Hepatitis B virus (HBV) is a small enveloped DNA virus belonging to the Hepadnaviridae family [1]. The structure of the HBV virion is as follows: 3.2 kb of partially double-stranded relaxed circular DNA (rcDNA) bonded by polymerase, an inner nucleocapsid formed by the core protein (hepatitis B core antigen, HBcAg), and an outer envelope formed by lipid-embedded small, middle, and large surface proteins (hepatitis B surface antigen, HBsAg). HBV is characterized by species specificity, and only humans and chimpanzees are susceptible [2]. One of the major factors determining species specificity is the interaction of the preS1 domain of large HBsAg with the sodium taurocholate cotransporting polypeptide (NTCP) of human hepatocytes (Fig. 1) [3].
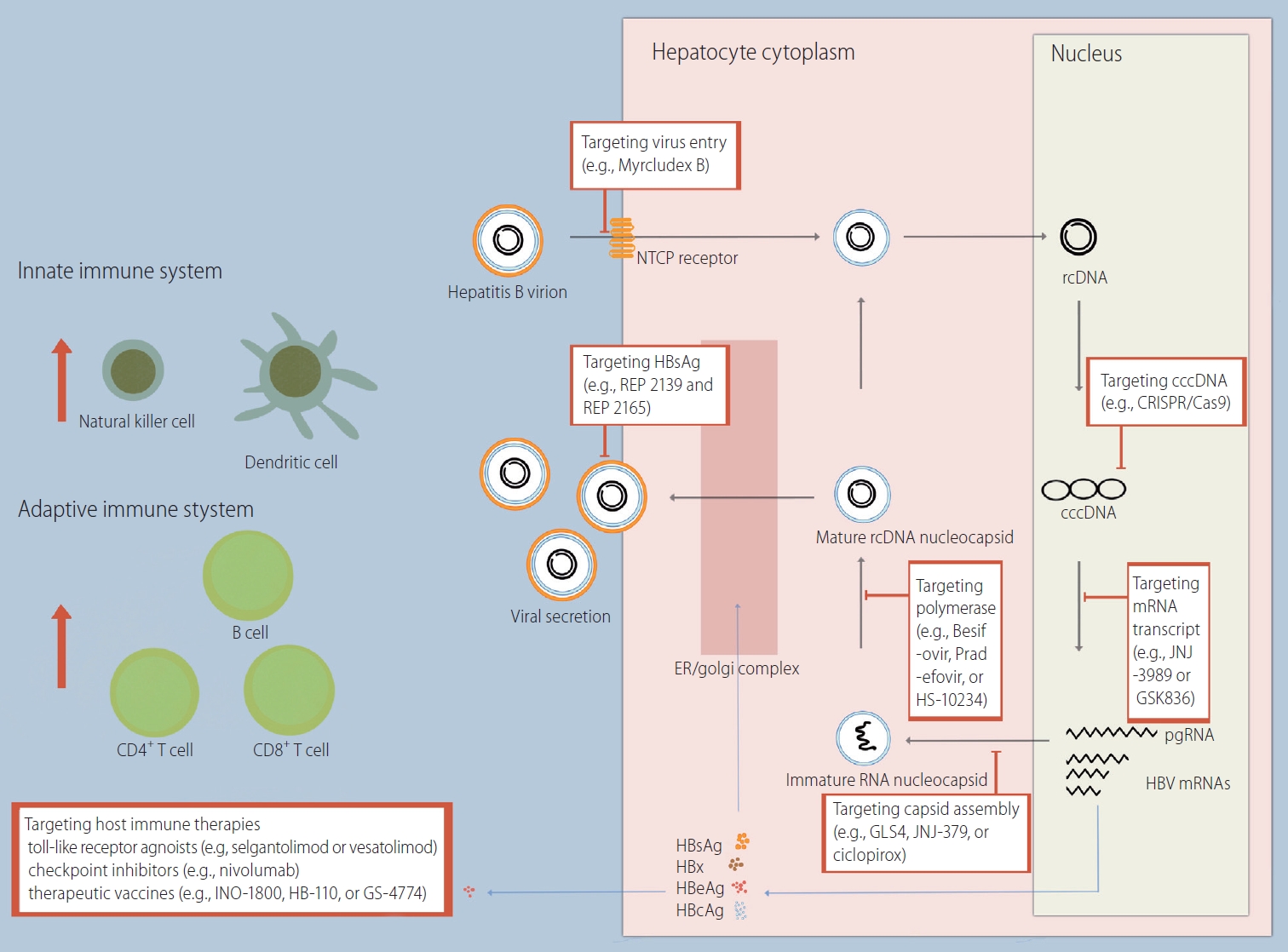
HBV life cycle and novel drug targets. NTCP, sodium taurocholate cotransporting polypeptide; rcDNA, relaxed circular DNA; cccDNA, covalently closed circular DNA; CRISPR, clustered regularly interspaced short palindromic repeats; Cas9, CRISPR-associated protein 9; ER, endoplasmic reticulum; mRNA, messenger RNA; pgRNA, pregenomic RNA; HBV, hepatitis B virus; HBsAg, hepatitis B surface antigen; HBeAg, hepatitis B envelope antigen; HBcAg, hepatitis B core antigen.
HBV virions enter the hepatocyte cytoplasm through a specific interaction with NTCP on the basolateral membrane of hepatocytes, followed by a plasma membrane component-mediated pathway (e.g., clathrin adaptor protein AP-2 and caveolin-1) [4,5]. Subsequently, translocation of HBV occurs using the endosomal network in the cytoplasm. The gradual decrease in the pH of the endosome (i.e., 6.2 in early endosomes and 5.5 in late endosomes) aids in the fusion of the endosomal membrane with the outer envelope proteins of HBV [6,7]. HBV DNA enters the nucleus after the inner nucleocapsid of HBV is disassembled by the nuclear pore complex.
In the nucleus, HBV DNA is converted from rcDNA to covalently closed circular DNA (cccDNA) [8]. cccDNA serves as a template for HBV gene products, including pregenomic RNA (pgRNA), preS1 messenger RNA (mRNA), preS2 mRNA, and X mRNA. The resulting RNAs are released into the cytoplasm. Some of the pgRNA serves as a translation template for the synthesis of the core protein and the polymerase. Another part of the pgRNA, together with the polymerase, forms a capsid using HBcAgs (i.e., encapsidation) [9,10]. Inside the capsid, pgRNA is reverse transcribed to rcDNA. PreS1 mRNA, preS2 mRNA, and X mRNA are used to synthesize surface proteins and HBx proteins. Newly synthesized HBV nucleocapsids are either used for intracellular cccDNA amplification or form HBV virions through assembly with surface proteins in the endoplasmic reticulum and are then released to the sinusoidal space. Surface proteins are also released in the form of noninfectious excess subviral particles (SVPs) that do not contain HBV DNA [11].
The treatment goals for patients with chronic hepatitis B (CHB) are to reduce the mortality rate from chronic liver disease and improve overall survival by preventing the development of cirrhosis and/or hepatocellular carcinoma (HCC). The recommended treatment strategies are as follows: 1) timely initiation of antiviral therapy for patients with CHB; 2) long-term suppression of HBV DNA levels; and 3) HBsAg loss, with or without anti-HBs seroconversion (i.e., a functional cure) as an optimal endpoint [12-14]. Note that a functional cure other than eradication of intrahepatic cccDNA and integrated HBV DNA (i.e., a complete cure) is the endpoint of the latter strategy. This endpoint reflects the therapeutic effects and limitations of the recommended first-line antiviral agents: nucleos(t)ide analogues (NA) and pegylated interferon alpha (Peg-INFα).
NA treatment has shown that over 60% of HBV DNA is undetectable after 48 weeks of treatment [15,16]. The effectiveness of NA treatment has been maintained for over 5 years [17,18]. However, the proportion of patients achieving HBsAg loss is only 1.4–3.0% at 48 weeks of treatment. Even NAs have little effect on cccDNA in nature [19]. Peg-INFα affects the entire replication process of HBV and can also induce degradation of cccDNA [20]. The rate of HBsAg loss exceeds 5% at 48 weeks of treatment [21,22]. However, Peg-INFα is difficult to use because the frequency of side effects is very high. At 48 weeks of treatment, alopecia, skin manifestations, insomnia, anxiety, and oral symptoms have been reported in 44.0%, 56.0%, 36.0%, 28.9%, and 64.0% of patients, respectively [23].
Although the risk of CHB-related HCC has been reduced as a result of NA or Peg-INFα treatment, the risk of HCC remains in patients with CHB [24-27]. CHB is still the leading cause of HCC, causing 44% of HCC cases worldwide and 65–75% of HCC cases in endemic regions like Korea [13,28]. To overcome the limitations of NAs and Peg-INFα and to achieve a complete cure for CHB, the need for drugs targeting novel therapeutic targets of HBV has emerged.
NOVLE THERAPEUTICS TARGETING VIRAL LIFE CYCLE
Targeting virus entry
Strategies to inhibit HBV entry include novel peptides derived from HBsAg, such as myrcludex B, a myristoylated peptide derived from 47 amino acids within the pre-S1 domain of L-HBsAg [29]. A multicenter, open-label phase 2 clinical trial was performed to evaluate the efficacy and safety of myrcludex B in combination with Peg-IFN2α for HBV/hepatitis D virus coinfection (Table 1) [30]. When myrcludex B was used as a monotherapy for 48 weeks, none of the patients experienced a HBsAg >1 log decline, and HBsAg was undetectable. When myrcludex B was used in combination with Pge-IFN2α over 48 weeks, HBsAg decline was achieved in 46.7% of the subjects (seven out of 15), indicating a potential role for myrcludex B in future HBV curative regimens.
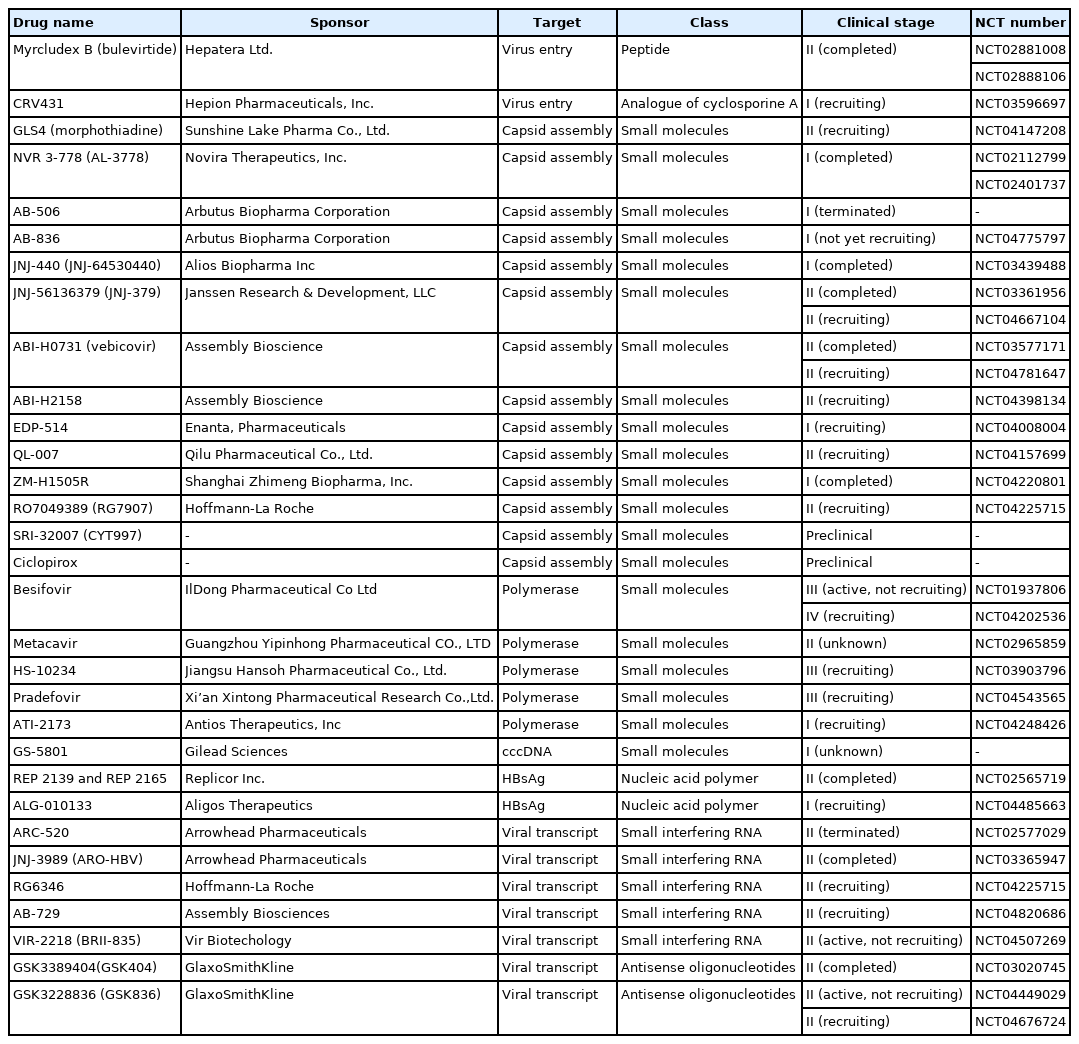
Selected novel agents in development for the treatment of chronic hepatitis B
Small molecules, such as ezetimibe [31], cyclosporine derivatives [32], and contravir [33], have also been evaluated as viral entry inhibitors in many experimental models, and some compounds inhibit entry independently of the NTCP receptor. A phase 1 single ascending dose study for contravir (CRV431) is currently recruiting study subjects to evaluate the safety and pharmacokinetics of CRV431 in combination with TDF.
Monoclonal antibodies against HBsAg epitopes (e.g., HBV-ABXTL, HepeX-BTM, and 2H5-A14) have been evaluated for their efficacy in blocking HBV entry into hepatocytes and/or accelerating viral clearance from the circulation. These antibodies may have additional antiviral function, as they could confer protection by binding circulating virus [34-37]. However, no clinical trials for monoclonal antibodies against HBsAg are ongoing.
Targeting capsid assembly
HBV capsid assembly plays a significant role in almost every step of the HBV replication cycle [38]. Notably, the HBV capsid is responsible for trafficking rcDNA to the nucleus, thereby establishing and sustaining cccDNA levels. Moreover, capsid protein is found in hepatocyte nuclei and interacts with host factors that are responsible for transcriptional regulation [9]. Therefore, targeting disruption of the HBV capsid protein could impact cccDNA stability and possibly lead to a complete cure of HBV infection [39]. Because sustained antiviral activity is expected, several capsid assembly modulators have been studied, such as GLS4 (phase 2 clinical trials) [40], NVR 3-778 (phase 2a clinical trials) [41], AB-423 (phase 1 clinical trials) [42], AB-506 (phase 1 clinical trials), JNJ-440 (phase 1 clinical trial) [43], JNJ-56136379 (phase 2 clinical trial) [44], ABI-H0731 (phase 2 clinical trials) [45], ABI-H2158 (phase 2a clinical trials), RO7049389 (phase 2 clinical trials) [46], Bay41-4109 (phase 1 clinical trials) [47], and QL-007 (phase 2 clinical trials). Structurally, these compounds are heteroaryldihydropyrimidines, phenylpropenamides, or sulfamoylbenzamides. Recently, a tubulin polymerization inhibitor (SRI-32007) was discovered as an inhibitor of HBV core promoter activity, and preclinical studies have been performed [48].
Recently, our research group discovered a novel anti-HBV drug, ciclopirox, from U.S. Food and Drug Administration-approved library [49]. Ciclopirox, a synthetic antifungal agent, strongly inhibited HBV replication in cells and mice by blocking assembly of the HBV capsid. The crystal structure of the HBV core protein and the ciclopirox complex revealed a novel binding mode at dimer-dimer interfaces. We also found that ciclopirox synergized with NAs to prevent HBV replication in cells and in a humanized liver mouse model. Orally administered ciclopirox may block HBV capsid assembly effectively and thus provide a novel opportunity to combat chronic HBV infection. Preclinical studies are ongoing for ciclopirox as a novel HBV drug.
Targeting polymerase
NAs, which are commonly used currently, are antiviral agents that target polymerase. Drugs targeting polymerase and that aim to improve the therapeutic effects or to reduce side effects better than NAs are being developed. Besifovir dipivoxil maleate (BSV) is an acyclic nucleotide phosphonate inhibitor of HBV polymerase [50]. In a phase 3 clinical study, BSV showed comparable efficacy and better safety compared to tenofovir disoproxil fumarate (TDF) [51]. Both HS-10234 and pradefovir, which are prodrugs of tenofovir and adefovir, respectively, showed comparable efficacy to TDF in a phase 2 clinical study and are in a phase 3 clinical study [52]. Metacavir, a novel deoxyguanosine analog, is under investigation in a phase 2 clinical trial [53]. ATI-2173 is a new chemical derived from clevudine, and it has the advantage of reducing the risk of clevudine-related myopathy. Currently, a phase 1 clinical trial is in progress.
Targeting cccDNA
Genome editing tools include zinc finger nuclease (ZFN), transcription activator-like effector nuclease (TALENs), and the clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) system [54,55]. The domains that recognize and bind to target double-stranded DNA (dsDNA) sequences in ZFN and TALENs are zinc finger protein and transcription activator-like effector, respectively [56,57]. Both tools require the FokI restriction endonuclease domain to cut the genome. On the contrary, all CRISPR/Cas9 systems use CRISPR RNA (i.e., guide RNA) to find target dsDNA and have a simple working structure that cuts target dsDNA with Cas protein endonuclease [58]. Among these tools, the CRISPR/Cas9 system is the most simplified tool for the development of custom nucleases. In a preclinical study, CRISPR/Cas9 system treatment showed encouraging results, showing that more than 90% of HBV DNA was cleaved by Cas9 [59,60]. Subsequently, the removal of the full-length integrated HBV DNA (i.e., sterile eradication) was also successful [61]. Nevertheless, the major obstacle to the clinical application of CRISPR/Cas9 is the possibility of expressing an off-target effect that causes damage to the host DNA rather than the target. As an alternative method, CRISPR/Cas9 nickase only causes a singlestrand DNA break, rather than dsDNA breaks [62]. Thus, a pair of properly spaced guide RNA can cause dsDNA breaks and the risk of permanent DNA damage due to off-target effects can be reduced.
In the nucleus, instead of performing genome editing, there are drugs that aim to silence cccDNA transcription. GS-5801 is a prodrug of a small molecule inhibitor of histone lysine demethylase 5 [63]. However, GS-5801 did not show antiviral activity in a humanized mouse model of HBV infection [64]. Other drugs that have blocked cccDNA transcription in the discovery stage are alpha-glucosidase, which suppresses the nuclear transcription factor Sp1 [65], and dicoumarol, which is a competitive nicotinamide adenine dinucleotide phosphate quinone oxidoreductase that acts as an inhibitor of HBx expression [66]. Peg-IFNα is involved in both the degradation and transcription of cccDNA [20,67]. Peg-INFα induces degradation of cccDNA through upregulation of APOBEC3A and 3B cytidine deaminases. Peg-INFα also blocks cccDNA transcription by inducing cccDNA-bound histone hypoacetylation.
Targeting HBsAg
Nucleic acid polymers (NAPs) have broad spectrum antiviral activity against several enveloped viruses, including HBV [68]. REP 2139 and REP 2165 are NAPs that inhibit the assembly and secretion of HBV SVP. In a phase 2 clinical trial, 48-week combination treatment with TDF, Peg-INFα, and NAP showed HBsAg loss in 60% of the participants; The functional cure persisted in 35% of the participants during 48 weeks of treatment-free follow up [69].
Targeting viral transcript
High levels of viral antigens may be associated with a suppressed host immune response and increased persistence of HBV. The synthesis of viral antigens could potentially be blocked by the use of molecular approaches targeting viral mRNA transcripts, such as RNA interference (RNAi) and antisense oligonucleotides.
Small interfering RNA (siRNA) acts as RNAi, which can target a common 3’ terminus shared by all mRNA transcripts and induce the degradation of the transcripts [70]. The degradation of mRNA transcripts can allow the suppression of HBV DNA and viral antigens, including HBsAg, HBeAg, and hepatitis B core-related antigens. Therefore, siRNA can reverse host immune exhaustion caused by high antigenemia, allow restoration of effective HBV-specific immune responses, and finally lead to the seroconversion of HBsAg. ARC-521 was the first siRNA against HBV designed to reduce all mRNA transcripts derived from cccDNA to be investigated in clinical trials [71]. The NA-experienced patients with CHB received four doses of ARC-520 at 1 mg/kg or 2 mg/kg or placebo every 4 weeks. The high-dose group (2 mg/kg) showed a significant reduction in HBsAg levels compared to the placebo group, but absolute HBsAg reductions were moderate in both patients who were hepatitis B envelope antigen (HBeAg)-negative (0.38 log IU/mL) and HBeAg-positive (0.54 log IU/mL). The limited activity was probably due to the high expression level of HBsAg from integrated HBV DNA. Unlike ARC-520, JNJ-3989 (ARO-HBV) can target viral mRNA transcripts from all sources, including cccDNA and integrated HBV DNA. In recent studies, 3 subcutaneous doses of JNJ-3989 were administered monthly in CHB patients concomitantly with entecavir or tenofovir [72]. JNJ-3989 with NA therapy was well tolerated and showed a ≥1.0 log IU/mL reduction in HBsAg in 98% of the patients with CHB.
Antisense oligonucleotides are nucleic acids that are designed to hybridize with target RNAs and block the expression of viral proteins. GSK3389404 is a liver-directed antisense oligonucleotide that inhibits the synthesis of viral proteins, including HBsAg. In a phase 2a clinical trial, subcutaneous injection of GSK3389404 (120 mg/week) for 3 months caused a rapid drop in the levels of HBsAg (0.75 log IU/mL) in patients with CHB [73].
Major concerns about the agents targeting viral transcripts include the potential toxicity of the delivery platform, the risk of off-target toxicity, and the risk of rebound after treatment due to a lack of cccDNA reduction.
HOST IMMUNE TARGETING THERAPIES
Adaptive immunity, such as HBV-specific T cell responses, is critical for the clearance of HBV infection. HBV-specific T cells secrete cytokines that induce noncytolytic clearance of HBV and recruitment of other inflammatory immune cells [74]. The innate immune response, such as the production of type I IFNs, is essential for the induction of following HBV-specific immunity. Therefore, the coordination of both innate and adaptive immune responses is important to control HBV infection. Patients with CHB are characterized by a weak innate and adaptive (HBV-specific) immune response [75]. Chronic HBV infection induces impaired cytokine production, T-cell exhaustion characterized by poor cytotoxic activity, and sustained expression of inhibitory receptors [76]. Immunomodulatory therapies focused on the restoration of impaired immune responses have been investigated in several trials as follows.
Toll-like receptor (TLR) agonists
TLRs are a type of pattern recognition receptor on eukaryotic cells that sense pathogen-associated molecular patterns leading to the production of antiviral mediators [77]. Therefore, TLRs are part of the innate immune system that constitutes the first line of defense against invading microorganisms. TLRs also play a pivotal role in adaptive immune responses by inducing the differentiation of naïve T cells into effector T cells. The activation of TLRs can suppress the replication of HBV and induce the restoration of HBV-specific adaptive immune responses. Vesatolimod (GS-9620), an oral agonist of TLR-7, activates innate and adaptive immune responses of patients with CHB. In a phase 2 clinical trial, once-weekly oral administration of vesatolimod for 4, 8, or 12 weeks showed a consistent dose-dependent induction of interferon-stimulated gene 15, which is the most sensitive indicator of TLR-7 activation (Table 2) [78]. However, vesatolimod therapy did not show a significant decline in HBsAg compared to placebo. Selgantolimod (GS-9688), an oral agonist of TLR-8, induced a sustained antiviral response in woodchuck hepatitis virus (a hepadnavirus that is closely related to HBV)-infected woodchucks [79]. In a phase 2 clinical trial, a combination of selgantolimod (up to 3 mg) once weekly and NA therapy for 24 weeks followed by NA therapy alone for an additional 24 weeks achieved a modest decline in HBsAg levels from baseline and a 5% rate of HBsAg loss [80].

Selected novel immune modulators in development for the treatment of chronic hepatitis B
Similar to TLR agonist, retinoic acid-inducible gene-I (RIG-I) agonist is another compound that can suppress the replication of HBV by boosting innate immune responses. RIG-I agonist boosts innate immunity directly by activating the intracellular interferon pathway in hepatocytes and also have a direct antiviral effect as a non-nucleotide reverse transcriptase inhibitor [81]. In a phase 2 clinical trial, a combination of inarigivir (SB9200), an oral dinucleotide RIG-I agonist, and tenofovir showed a dose-dependent reduction of HBV DNA [82]. However, further investigation of inarigivir has been stopped due to severe adverse events including the death of one patient in another phase 2 clinical trial of the use of inarigivir 400 mg plus NA in HBeAg-negative patients [83].
Checkpoint Inhibitors
In chronic HBV infection, upregulation of checkpoint inhibitors, such as programmed cell death protein 1 (PD-1), is associated with T-cell exhaustion and persistent viral infection. Therefore, checkpoint inhibitors may help to restore T cell function. The induction of uncontrolled hepatitis flares and autoimmunity are the main concerns of this type of therapy. In a phase 1b clinical trial, nivolumab, a PD-1 inhibitor, was administered at either 0.1 or 0.3 mg/kg with or without a HBV therapeutic vaccine (GS-4774) in virally suppressed patients with HBeAg-negative CHB [84]. Patients receiving 0.3 mg/kg of nivolumab with and without GS-4774 at baseline and week 4 showed significant HBsAg declines at week 24 from baseline, and one patient receiving the combination experienced HBsAg loss. There were no serious adverse events. Other checkpoint inhibitors targeting cytotoxic T lymphocyte-associated antigen-5 and CD244/2B4 restored immune function and increased the proliferation of CD8 T cells in vitro [85,86].
Therapeutic vaccines
Therapeutic vaccination aims to restore HBV-specific immunity by stimulating the host immune response. In previous trials, existing prophylactic vaccines were able to reduce HBV replication in animal models of chronic hepadnaviral infection [87] but failed in patients with CHB [88,89]. These vaccines induced antibodies but failed to induce cytotoxic T cell responses that would be critical for therapeutic efficacy. Alternatively, DNA vaccines coding sequences of HBsAg to induce HBV-specific T cells were investigated but also yielded poor results [90,91]. DNA vaccines against multiple HBV proteins employing heterologous prime-boost regimens are currently under investigation. INO-1800, which is a DNA-based HBV vaccine encoding HBsAg and HBcAg, induced the binding of antibodies to HBs and robust cell-mediated immunity in animals [92]. The use of INO-1800 with or without human interleukin 12 in CHB patients virally suppressed with a NA is in a phase 1 clinical trial (NCT02431312). Combined treatment with HB-110, a DNA vaccine encoding S, L, core, and polymerase proteins, and adefovir was also investigated in a phase 1 clinical trial [93]. HB-110 exhibited a weak capability to induce HBV-specific T cell responses. Other HBV vaccines encoding multiple HBV proteins produced by different vectors, including GS-4774 and TG-1050, have been demonstrated to induce immunogenicity in animal models and healthy individuals [94,95]. However, in a clinical trial, there were no significant differences in the decline in HBsAg levels between the GS4774 group and the tenofovir group [94].
Genetically engineered T cells
To restore adequate HBV-specific T cell immunity, the adoptive transfer of genetically engineered T cells can be a promising strategy [75]. T cells separated from the peripheral blood of patients with CHB were expanded and activated in vitro. The expanded T cells were engineered to encode HBV-specific T cell receptors by using viral vectors and then were reinfused to patients with CHB. This approach could be achieved through T-cell receptor (TCR) gene transfer or the use of chimeric antigen receptor (CAR) T cells [96]. TCR-redirected HBV-specific T cells were able to recognize HBV-infected cells in an in vitro study [97]. T cells with a CAR specific for HBeAg were also reported to localize to the liver of mice to reduce the replication of HBV with only transient liver damage [98].
NEEDS FOR COMBINATION TREATMENT
We reviewed some of the anti-HBV agents targeting virus entry, capsid assembly, polymerase, cccDNA, HBsAg, viral transcripts, or HBV-specific immune responses (immunomodulators). Considering the mechanism of HBV replication and the difficulty in eradicating cccDNA and integrated HBV DNA, combination treatment using two or more drugs with different targets may be more advantageous than monotreatment.
The results of combination treatment with NAs and Peg-INFα differed depending on the type of NA. Lamivudine and Peg-INFα combination treatment was inferior to monotherapy [99-101]. However, TDF and Peg-INFα combination treatment showed a significantly higher proportion of HBsAg loss than monotreatment with TDF or Peg-INFα after 72 weeks [102]. Recently, a randomized trial found that combination treatment with entecavir (ETV), PEG-INFα, and sequential vaccination showed a significantly higher proportion of HBsAg loss than monotreatment with ETV after 100 weeks (16.2% vs. 0.0%) [103].
The therapeutic effects of combined treatment with a NA and novel immunomodulators have also been studied. Combination treatment with a NA and GS-4774 did not further reduce HBsAg levels compared to monotreatment with NA in a phase 2 clinical trial [94]. Combination treatment with ETV and thymosin alpha-1 did not significantly improve the rate of undetectable HBV DNA or the rate of HBsAg loss after 52 weeks compared to mono ETV treatment [104]. However, in a phase 2 clinical trial, combination treatment with a NA and selgantolimod showed a 5% rate of HBsAg loss after 24 weeks [80]. Similarly, HBsAg loss was observed in 25% of patients at 4 weeks of treatment in a phase 2a clinical trial of NA and GSK3228836 combination treatment [105]. Currently, a phase 2 clinical trial is ongoing to evaluate the efficacy and safety of the combined treatment of NA, GSK3228836, and Peg-INFα. Therefore, the combination of direct-acting antivirals and novel immunomodulators might be the ideal strategy to completely cure patients infected by HBV.
ENDPOINTS OF CLINICAL TRIALS
The main endpoint of recent phase 2/3 clinical trials of novel HBV therapy was the functional cure of HBV (i.e., HBsAg loss) and therefore, quantitative HBsAg is a useful marker for assessing the efficacy of the novel therapy. However, HBsAg is not an ideal marker for very early assessment of the clinical trials because HBsAg may be expressed from cccDNA and integrated HBV DNA. Therefore, circulating viral RNAs or hepatitis B core-related antigen, potential surrogate markers of cccDNA, are also important markers for assessing the novel therapy. In the future clinical trials of novel agents targeting cccDNA elimination, the achievement of complete cure of HBV (i.e, elimination of cccDNA and integrated HBV DNA) will be an important endpoint. Unlike the ease of judgment of the functional cure, the standard evaluation method of the complete cure has not yet been established. Liver biopsy may be used to determine cccDNA elimination [19]. However, it is challenging to accurately determine the absence of cccDNA due to detection limitations [106].
The appropriate time point for evaluating the treatment response for the phase 3 clinical trial suggested by the expert group was 6 months after the end of treatment [107]. It was agreed that it was an appropriate time to determine if the treatment response persisted. However, in the case of the phase 2 clinical trial, a specific time point was not presented. Considering the mechanism of the novel drug, the evaluation time point can be altered as 6 months after the end of treatment or 6 months after the start of treatment.
FUTURE DIRECTIONS
The treatment options introduced in this study have different mechanisms of action, and each option has strengths and potential limitations. Therefore, combination therapy is promising for achieving a functional cure for HBV infection. Novel curative agents for HBV can be stratified either as direct regulators of the HBV life cycle (direct-acting antivirals) or indirect regulators of innate and/or HBV-specific immunomodulators. Combination treatment with potent NAs that can effectively suppress viral replication, one or more direct-acting antiviral, and at least one immunomodulator can be a promising approach for treating chronic HBV infection. In addition, treatments should be individualized by considering different host factors, including treatment history, HBeAg expression, viral load, severity of fibrosis, and genotype. Future trials combining multiple therapeutic agents considering baseline host factors are warranted to find optimal and customized regimens for patients with CHB.
CONCLUSION
The key obstacle to achieving a complete cure for HBV infection is the presence of cccDNA in the hepatocyte nucleus. The cccDNA acts as a transcription template for all viral RNAs, including pgRNA, and has a very long half-life. The removal of cccDNA is very difficult because cccDNA resides in the nucleus as a stable episomal plasmid-like molecule. Nevertheless, recent advances in technology and expanded knowledge about HBV infection have led to the development of agents targeting cccDNA. In addition, other direct-acting antivirals that can target various steps of the HBV life cycle, including HBV entry, transcription of viral proteins, nucleocapsid assembly, pgRNA packaging, and HBsAg release, are currently under investigation in clinical trials. Another barrier to finding a complete cure for HBV infection is defective host immune responses. Chronic HBV infection induces weak innate and HBV-specific immune responses, such as impaired cytokine production, T-cell exhaustion, and sustained expression of inhibitory receptors. Immunomodulators targeting innate and/or HBV-specific immune responses, including TLR agonists, checkpoint inhibitors, therapeutic vaccines, and engineered T cells, are also currently under development or in clinical trials. The combination of novel agents with or without potent NAs may be a promising strategy for a functional cure of HBV infection. Continued advancements of the novel agents may aid in the elimination of HBV.
Notes
Authors’ contribution
SW Kim, JS Yoon, Y Cho wrote the paper, M Lee and Y Cho supervised the whole project, and Y Cho edited the paper.
Conflicts of Interest: The authors have no conflicts to disclose.
Acknowledgements
This research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute, funded by the Ministry of Health & Welfare, Republic of Korea (No. HI21C0240), and the National Research Foundation of Korea grant funded by the Korea government (No. 2021R1A2C4001401).
Abbreviations
BSV
Besifovir dipivoxil maleate
CAR
chimeric antigen receptor
Cas9
CRISPR-associated protein 9
cccDNA
covalently closed circular DNA
CHB
chronic hepatitis B
CRISPR
clustered regularly interspaced short palindromic repeats
dsDNA
double-stranded DNA
EVT
entecavir
HBcAg
hepatitis B core antigen
HBeAg
hepatitis B envelope antigen
HBsAg
hepatitis B surface antigen
HBV
hepatitis B virus
HCC
hepatocellular carcinoma
mRNA
messenger RNA
NA
nucleos(t)ide analogues
NAP
nucleic acid polymer
NTCP
sodium taurocholate cotransporting polypeptide
PD-1
programmed cell death protein 1
Peg-INFα
pegylated interferon alpha
pgRNA
pregenomic RNA
rcDNA
relaxed circular DNA
RIG-I
retinoic acid-inducible gene-I
RNAi
RNA interference
siRNA
small interfering RNA
SVP
subviral particle
TALENs
transcription activator-like effector nuclease
TCR
T-cell receptor
TDF
tenofovir disoproxil fumarate
TLR
toll-like receptor
ZFN
zinc finger nuclease