Anti-fibrotic treatments for chronic liver diseases: The present and the future
Article information

Abstract
Liver fibrosis reflects tissue scarring in the liver due to the accumulation of excessive extracellular matrix in response to chronically persistent liver injury. Hepatocyte cell death can trigger capillarization of liver sinusoidal endothelial cells, stimulation of immune cells including macrophages and Kupffer cells, and activation of hepatic stellate cells (HSCs), resulting in progression of liver fibrosis. Liver cirrhosis is the terminal state of liver fibrosis and is associated with severe complications, such as liver failure, portal hypertension, and liver cancer. Nevertheless, effective therapy for cirrhosis has not yet been established, and liver transplantation is the only radical treatment for severe cases. Studies investigating HSC activation and regulation of collagen production in the liver have made breakthroughs in recent decades that have advanced the knowledge regarding liver fibrosis pathophysiology. In this review, we summarize molecular mechanisms of liver fibrosis and discuss the development of novel anti-fibrotic therapies.
INTRODUCTION
Liver cirrhosis is a late-stage of chronic hepatitis and currently the 11th most common cause of death globally [1]. Decompensated cirrhosis, the most advanced stage of cirrhosis, is accompanied by severe complications, including liver failure, opportunistic infection, and portal hypertension (resulting in ascites, hepatic encephalopathy, or gastroesophageal varices), that threaten the lives of patients [2]. Cirrhosis is accompanied by extensive tissue scarring and an increase in intrahepatic vascular resistance. Cirrhosis develops from chronic hepatitis, that can be caused by hepatitis B virus (HBV), hepatitis C virus (HCV), alcoholic liver disease (ALD), non-alcoholic steatohepatitis (NASH), autoimmune hepatitis, and genetic diseases, including hemochromatosis and Wilson’s disease [3]. Recently, progress made in antiviral drugs has contributed to a decrease in viral hepatitis, while the proportion of cirrhosis and liver cancer caused by ALD and NASH has been increasing, particularly in western countries [4]. Research on liver fibrosis, including the development of cirrhosis therapy, has made remarkable progress. However, effective drugs for cirrhosis treatment are not yet available for clinical use. Development of effective cirrhosis therapies requires the ability to not only target specific cell types, but also to elucidate further mechanisms of liver fibrosis with a comprehensive understanding of intercellular molecular networks. This review will highlight the current status of anti-fibrotic drug development and review the recent studies investigating the molecular mechanisms of liver fibrosis.
POSSIBILITY OF CELL-TARGETING STRATEGY FOR ANTI-FIBROTIC DRUGS
Liver fibrosis is the most common pathology of cirrhosis and is characterized by progressive accumulation of extracellular matrix (ECM), which destroys the lobule architecture of the liver [5]. Most of the liver injury is associated with hepatocyte damage. Liver injury induces the accelerated production of ECM components, such as collagens, elastin, and proteoglycans [6]. This bioadaptive reaction protects hepatocytes from cell death and contributes to liver regeneration. However, persistent liver injury increases activated hepatic stellate cells (HSCs) that produce excessive ECM components, primarily type 1 collagen encoded by the COL1A1 and COL1A2 genes [7]. Since the amount of type I collagen in liver tissues results from the equilibrium between type 1 collagen production and the activities of matrix metalloproteinases (MMPs) and tissue inhibitor of MMPs, disruption of the equilibrium can lead to progression of liver fibrosis [8,9]. In addition, upon liver injury, bone marrow-derived inflammatory cells, including macrophages, accelerate HSC activation with high production of type I collagen. Therefore, strategies for liver fibrosis therapy include: 1) inhibition of HSC activation; 2) reduction of fibrotic scar development; 3) immune modulation; and 4) protection from hepatocyte death (Fig. 1).

Cell-targeting strategy for anti-fibrotic therapy. Chronic liver injury due to various etiologies causes hepatocyte damage and hepatocyte death. In response to persistent hepatocyte damage, activation of HSCs and macrophages (including Kupffer cells) is induced. Enhanced by activated macrophages, activated HSCs increase and produce excessive ECM, resulting in progression of liver fibrosis. Four major strategies can be utilized to target the interplay between hepatocytes, HSCs, and immune cells involved in the molecular mechanisms associated with collagen accumulation can: 1) inhibition of HSC activation; 2) reduction of fibrotic scar evolution; 3) immune modulation; and 4) protection from hepatocyte death. Numerous drugs that incorporate these strategies are currently being evaluated in clinical trials. CCR, C-C chemokine receptor; HSC, hepatic stellate cell; PPARγ, peroxisome proliferator-activated receptor-γ; FXR, farnesoid X receptor; CBP, CREB-binding protein; NOX, NADPH oxidase; ECM, extracellular matrix; NASH, non-alcoholic steatohepatitis; ASK1, apoptosis signal-regulating kinase 1; FGF, fibroblast growth factor; SCD1, stearoyl-coenzyme A desaturase 1; THR-β, thyroid hormone receptor-β; FGF, fibroblast growth factor; HBV, hepatitis B virus; HCV, hepatitis C virus; DAAs, direct-acting antivirals; AIH, autoimmune hepatitis; PBC, primary biliary cholangitis; UDCA, ursodeoxycholic acid; LOXL2, lysyl oxidase-like 2; HSP47, heat shock protein 47.
Current therapeutic strategies for liver fibrosis rely primarily on the elimination of etiologies. In fact, clinical evidence for liver fibrosis resolution has emerged from studies investigating antiviral therapies for viral hepatitis [10-12], lifestyle changes, and bariatric surgery for metabolic liver disease [13], suggesting that liver fibrosis is indeed reversible. Table 1 illustrates a portion of the liver fibrosis clinical trials that are currently active or in recruiting phase 1–3. Here, developing anti-fibrotic drugs are summarized from the perspective of cell-targeting strategies.
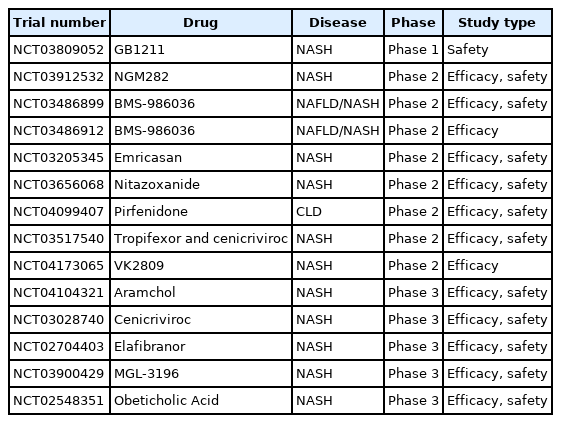
Current active or recruiting phase 1–3 clinical trials for liver fibrosis (ClinicalTrials.gov)
Inhibition of HSC activation
HSCs have a quiescent status in healthy liver tissue. Quiescent HSCs (qHSCs) store retinol as retinyl palmitate in lipid droplets. In response to liver injury, HSCs activate and transdifferentiate into myofibroblast-like cells [7,14]. Activated HSCs, characterized by decreased lipid droplets and enhanced expression of α-smooth muscle actin, have a proliferative phenotype and produce excessive ECM components, primarily collagens, resulting in progression of liver fibrosis [15,16]. HSCs are activated by cytokines, such as interleukins (ILs), tumor necrosis factor- alpha (TNF-α), transforming growth factor-β (TGF-β), platelet-derived growth factor, and chemokines, such as monocyte chemoattractant protein-1 (also known as C-C motif ligand 2 [CCL2]), and C-X-C motif ligand 9. Hepatocyte cell death caused by chronic liver injury causes inflammatory activity of liver immune cells, predominantly macrophages, leading to an increase in these secretory factors [17]. In addition, pathogen-associated molecular patterns (PAMPs) such as lipopolysaccharides released from microbes, DNA, and damaged-associated molecular patterns (DAMPs) derived from damaged hepatocytes, can activate HSCs. PAMPs and DAMPs bind to pattern recognition receptors, such as toll-like receptor 4, which then enhances the nuclear translocation of nuclear factor-kappa B (NF-κB) via myeloid differentiation primary response 88, and downregulates the expression of BMP and activin membrane-bound inhibitor, thereby restricting TGF-β signaling [18,19].
Peroxisome proliferator-activated receptors (PPARs) are ligand-activated transcription factors of the nuclear hormone receptor superfamily that play major regulatory roles in energy homeostasis and metabolic function. Among the three isoforms of PPARs (PPARα, PPARγ, and PPARβ/δ), PPARγ is especially considered as a promising therapeutic target of liver fibrosis. The expression of PPARγ decreases HSC activation, though it is highly expressed in quiescent HSCs [20]. A randomized phase 3 clinical trial of pioglitazone, a synthetic insulin sensitizing PPARγ agonist, reported improved steatosis and lobular inflammation without a significant effect on fibrosis, in non-cirrhotic NASH patients [21]. However, a subsequent meta-analysis of eight randomized clinical trials for thiazolidinedione therapy showed that pioglitazone treatment, for up to 24 months, was associated with fibrosis improvement at any stage and NASH resolution [22]. Moreover, elafibranor is a dual PPARα/δ agonist that has shown liver protective effects for steatosis, inflammation, and fibrosis in murine models of non-alcoholic fatty liver disease (NAFLD)/NASH and liver fibrosis [23]. Results from a post-hoc analysis from a phase 2 trial (NCT01694849) for elafibranor treatment (120 mg/day for 1 year) suggest that it resolved NASH without worsening fibrosis in patients with moderate to severe NASH [24]. Further, a phase 3 trial (NCT02704403) is currently underway in NASH patients without cirrhosis, with an endpoint defined as resolution of NASH without worsening of fibrosis. However, according to an interim analysis of this trial, elafibranor showed no significant effect on NASH resolution without worsening of fibrosis [25].
Farnesoid X receptor (FXR), which binds to bile acids as ligands, enhances insulin sensitivity and fatty acid beta-oxidation, while reducing gluconeogenesis and lipogenesis in hepatocytes [26]. FXR is highly expressed in the small intestine and liver and is also expressed in HSCs. Interestingly, overexpression of FXR inhibited production of collagen in HSCs [27]. Meanwhile, obeticholic acid (OCA), a semisynthetic derivative of chenodeoxycholic acid, is the selective FXR agonist. In a phase 2 trial, administration of 25 or 50 mg of OCA for 6 weeks increased insulin sensitivity, and reduced markers of liver inflammation and fibrosis in patients with type 2 diabetes mellitus and NAFLD (NCT00501592) [28]. Furthermore, an interim analysis of an ongoing phase 3 study in patients with NASH (NCT02548351) recently reported that daily administration of 25 mg of OCA significantly improved fibrosis by at least one stage without worsening NASH [29]. Though this report has suggested that NASH patients with non-cirrhotic advanced fibrosis might benefit from OCA treatment, the U.S. Food and Drug Administration (FDA) has not approved OCA for the treatment of NASH fibrosis and has requested the submission of additional post-interim analyses on the efficacy and safety of the ongoing study [30].
Wnt/β-catenin signaling is associated with the development of tissue fibrosis, including liver fibrosis [31]. PRI-724, a cyclic AMP-response element binding protein-binding protein (CBP)/β-catenin inhibitor, has been shown to inhibit HSC activation and collagen production in HCV transgenic mice [32]. Additionally, inhibition of CBP/β-catenin signaling reportedly attenuated liver fibrosis via reduced hepatocyte apoptosis and suppression of collagen-producing cell activation. According to a phase 1 study, intravenous administration of 10 or 40 mg/m2/day of PRI-724 over 12 weeks was well tolerated by patients with HCV cirrhosis showing dose dependent histological improvement (>2 points decrease in histologic activity index score) in 3/12 patients, and deterioration by 2 points in 2/12 patients [33]. A phase 1/2a clinical trial for PRI-724 in patients with hepatitis B or C related liver cirrhosis is ongoing.
Oxidative stress is a known cause of liver fibrosis progression, particularly in NASH [34]. NADPH oxidase (NOX) is a common source of reactive oxygen species (ROS) generation [35,36], and NOX1, 2, and 4 play important roles in the activation of HSCs [37,38]. GKT137831, a NOX1/4 inhibitor, suppresses ROS production, NOX, and fibrotic gene expression, while attenuating liver fibrosis in carbon tetrachloride (CCl4)-induced liver fibrosis in mice [39]. A phase 2 clinical trial for GKT137831 in patients with primary biliary cholangitis (PBC) (NCT03226067) has been completed and results are awaiting publication.
Nitazoxanide (NTZ), an antiprotozoal agent, is the only FDA-approved drug for Cryptosporidium infection [40]. Recently, NTZ was identified as a potent anti-fibrotic agent by a phenotypic screening approach aimed at discovering a compound capable of interfering with HSC activation. NTZ was found to reduce liver fibrosis in murine models of both CCl4-induced liver fibrosis and diet-induced NASH [41]. Additionally, NTZ, together with elafibranor, work synergistically to reduce liver fibrosis in a murine NASH model [42]. A phase 2 trial to evaluate the efficacy of NTZ in NASH patients with severe fibrosis is ongoing.
Cytoglobin (CYGB), the fourth human globin, discovered in our laboratory, is most abundantly expressed in HSCs among liver cells [43,44]. CYGB can bind with oxygen and nitric oxide, and is believed to protect HSCs from ROS [45]. TGF-β1-induced suppression of human CYGB expression is reported to contribute to the promotion of HSC activation via loss of cellular tolerance to exogenous oxidative stress and oxidative DNA damage in HSCs, resulting in acceleration of liver fibrosis [46]. Cygb-deficient mice presented with progressed liver fibrosis and susceptibility to liver cancer progression in diethylnitrosamine-induced hepatocellular carcinoma and NASH models [47]. Furthermore, in a rat model, CCl4-induced liver fibrosis was alleviated by administration of recombinant human CYGB protein [48]. Fibroblast growth factor 2, which enhances CYGB expression, attenuated the progression of liver fibrosis in mice with bile duct ligation [49]. CYGB protein may, therefore, represent a potential anti-fibrotic drug.
Removal of activated HSCs offers an alternative potential therapeutic strategy for liver fibrosis. Terminal deoxynucleotidyl transferase dUTP nick end labeling-positive HSCs are reportedly increased following reduced liver fibrosis during the recovery process from bile duct ligation-induced liver injury in rats [50]. Moreover, human and rodent liver myofibroblasts experience constitutive NF-κB activation, which promotes survival by inducing expression of anti-apoptotic genes, such as growth arrest and DNA-damage-inducible 45 beta and B-cell lymphoma 2 [51]. Therefore, studies on the effectiveness of targeted HSC apoptosis as a therapeutic strategy for liver fibrosis are being actively conducted [52,53].
Reduction of fibrotic scar evolution
Lysyl oxidase-like 2 (LOXL2) is a copper-dependent amine oxidase secreted by HSCs that contributes to liver fibrosis by catalyzing collagen cross-linking [54,55]. In murine models of fibrosis, inhibition of LOXL2 by an anti-LOXL2 murine monoclonal antibody decreased liver fibrosis and increased survival [54]. Meanwhile, simtuzumab, a humanized monoclonal antibody against LOXL2, inhibits the enzymatic activity of LOXL2 [56]. Unfortunately, in two phase 2b trials for NASH patients with bridging fibrosis or compensated cirrhosis (NCT01672866 and NCT01672879), simtuzumab failed to decrease hepatic collagen content or hepatic venous pressure gradient (HVPG), respectively [57]. Similarly, in a phase 2 study on primary sclerosing cholangitis patients (NCT01672853), treatment with simtuzumab for 96 weeks did not reduce fibrosis stage, progression to cirrhosis, or frequency of clinical events [58].
Collagen 1 accounts for the most abundant collagen in fibrotic livers [59]. A previous study demonstrated that lipid nanoparticles loaded with small interfering RNA (siRNA) against the procollagen α1(I) gene specifically reduced total hepatic collagen content in murine model of liver fibrosis [60]. Furthermore, another study using transgenic mice with inducible collagen 1 knockdown reported a 40–50% reduction in hepatic collagen accumulation with additional anti-inflammatory effects [61]. Meanwhile, heat shock protein 47 (HSP47) is a collagen-specific molecular chaperone essential for procollagen folding in the endoplasmic reticulum [62,63]. Sato et al. [64] reported that vitamin A-coupled liposomes carrying siRNA against mRNA encoding rat gp46, a homolog of HSP47, resolved liver fibrosis in a rat model of liver fibrosis. The efficacy of BMS-986263, an HSP47 siRNA delivering lipid nanoparticle, has be investigated in patients with F3–4 liver fibrosis (NCT02227459); the interim results for which have demonstrated that BMS-986263 was well tolerated and showed histologic improvement in fibrosis [65].
Immune modulation
Infiltrating inflammatory cells, particularly macrophages, are involved in liver fibrosis. PAMPs and DAMPs can stimulate the activation of Kupffer cells, resident macrophages in the liver, and induce immune and inflammatory reactions in the liver. Activated Kupffer cells not only promote HSC activation, but also secrete chemokines, including C-C chemokine ligand (CCL) types 2 and 5 (CCL2 and CCL5), which together with their respective receptors, C-C chemokine receptor (CCR) types 2 and 5 (CCR2 and CCR5), contribute to liver inflammation and fibrosis [66-70]. In response to liver injury, Kupffer cells secrete CCL2 and promote monocyte recruitment to the liver, followed by their maturation into pro-inflammatory LY6Chigh macrophages [71,72]. Pro-inflammatory cytokines released from the macrophages activate HSCs by promoting collagen production [73]. In fact, a dual CCR2/CCR5 inhibitor, cenicriviroc (CVC) reportedly reduced recruitment of pro-inflammatory macrophages and exerted anti-fibrotic effects in animal models of liver fibrosis [74,75]. Moreover, a phase 2b randomized study (NCT02217475) has reported that after 1 year of CVC treatment, twice as many subjects achieved an improvement in fibrosis without worsening of steatohepatitis, compared with placebo [76]. A rollover study using CVC for the treatment of liver fibrosis in NASH patients is ongoing (NCT03059446). Currently, another rollover study to assess the long-term safety of CVC is being conducted in patients with NASH who have participated in either the CENTAUR study (NCT02217475) or the AURORA study (NCT03028740). Additionally, a combination therapy comprising CVC and tropifexor (an FXR agonist), is under investigation and has reportedly improved inflammation and ballooning in an animal model of NASH. A phase 2 trial of this combination therapy in patients with NASH and liver fibrosis (F2 or 3) is ongoing [77].
Galectin-3 is primarily secreted by activated macrophages and is involved in the pathophysiology of liver fibrosis [78-80]. Previous studies have demonstrated that belapectin (also known as GRMD-02), an inhibitor or galectin-3, showed potent anti-fibrotic efficacy in mouse and rat models of liver fibrosis [81,82]. While a phase 2b study of belapectin (NCT02462967) did not elicit significant effects on fibrosis following treatment for 52 weeks in patients with NASH, cirrhosis, and portal hypertension, 2 mg/kg of belapectin effectively reduced HVPG and development of varices in a subgroup analysis of patients without esophageal varices [83]. A phase 2b/3 clinical study in patients with NASH cirrhosis without varices is ongoing (NCT04365868). Additionally, GB1211, another galectin-3 receptor inhibitor, is being investigated for its safety and tolerability in a phase 1 study (NCT03809052).
In addition, inflammasomes in hepatic macrophages are important players in liver fibrosis. A well-studied PRR, NLR family pyrin domain containing 3 (NLRP3), forms a complex referred to as the “NLRP3 inflammasome,” which produces and secretes inflammatory cytokines [84-86]. Calvente et al. [87] demonstrated that neutrophilderived microRNA-223 acts as a silencer of Nlrp3 in hepatic macrophages, resulting in attenuated fibrogenesis via inhibition of collagen synthesis in HSCs. Thus, macrophage-specific inhibition of inflammasomes may be a promising strategy for liver fibrosis therapeutics.
Protection from hepatocyte death
Preventing hepatocyte death by eliminating the cause of hepatocyte injury is one of the most essential treatment strategies for liver fibrosis. Recently, many new drugs to prevent hepatocyte death have been developed and tested, particularly for NASH. In this section, anti-fibrotic drugs targeting hepatocyte injury and death caused by NASH or other factors, are summarized.
NASH
Previous reports have shown that hepatocyte cell death induces liver inflammation and HSC activation, leading to liver fibrosis progression; hence, inhibition of hepatocyte death could decrease HSC activation in animal models [88,89]. Recently, randomized placebo-controlled trials for emricasan, a pan-caspase inhibitor, investigated its efficacy in NASH patients. Though emricasan slightly improved HVPG in cirrhotic NASH patients (NCT02960204) [90], it did not improve liver inflammation or fibrosis, but rather had a tendency to worsen hepatocyte ballooning in patients with NASHassociated F1–F3 fibrosis (NCT02686762) [91]. Furthermore, emricasan did not meet the primary endpoint in a phase 2b trial in patients with decompensated NASH cirrhosis (NCT03205345) [92].
Apoptosis signal-regulating kinase 1 (ASK1) is activated by various pathological stimuli, including intracellular oxidative stress and endoplasmic reticulum stress. Activation of ASK1 is involved in hepatocyte apoptosis and necrosis, leading to inflammation and fibrosis in the liver [93-95]. A selective ASK1 inhibitor, selonsertib, was investigated for NASH therapy in a phase 2 clinical trial, and demonstrated improved histological fibrosis in NASH patients with F2–3 fibrosis after 24 weeks of treatment [96]. However, randomized phase 3 trials in NASH patients with F3 (NCT03053050) and F4 fibrosis (NCT03053063) reported no significant anti-fibrotic effect after 48 weeks of selonsertib monotherapy [97].
TNF-α also induces hepatocyte death and acute liver failure [98,99]. Apoptotic bodies produced during hepatocyte death are engulfed by Kupffer cells, resulting in enhanced production of death ligands (TNF-α, TRAIL, and FasL) by Kupffer cells and further induction of hepatocyte death [100-102]. Pirfenidone (PFD), an orally bioavailable pyridone derivative, is approved for the treatment of idiopathic pulmonary fibrosis [103,104]. A previous study has reported that treatment with PFD for 24 months improved hepatic inflammation and fibrosis in patients with chronic hepatitis C [105]. However, the mechanism of action of PFD has not been fully elucidated. Nevertheless, a recent study reported that PFD attenuated liver fibrosis in western diet-fed melanocortin 4 receptor-deficient mice (NASH model mice). PFD also prevented TNF-α-induced hepatocyte apoptosis with reduced activation of caspase-8 and caspase-3, suggesting that PFD exerts anti-fibrotic effects in NASH via inhibition of hepatocyte death [106]. A phase 2 study (NCT04099407) evaluating the anti-fibrotic effect and safety of PFD treatment for 12 months in patients with chronic liver diseases has recently reported a significant reduction of fibrosis in 35% of PFD-treated patients [107].
BMS-986036 (Pegbelfermin) is a polyethylene glycol-conjugated recombinant analog of human fibroblast growth factor 21 [108,109], which is a hepatokine that regulates glucose and lipid metabolism in white adipose tissue [110]. According to the results of a phase 2 study in NASH patients (NCT02413372), pegbelfermin administration for 16 weeks (10 mg once per day, or 20 mg once per week) significantly reduced both hepatic fat fraction, as measured by magnetic resonance imaging-proton density fat fraction, and mean liver stiffness, as measured by magnetic resonance elastography, compared to the placebo group [111]. Phase 2 trials investigating the histologic effects of pegbelfermin are ongoing in NASH patients with bridging fibrosis (NCT03486899), as well as in those with NASH and compensated cirrhosis (NCT03486912), and is projected to be completed in 2021.
Statins which inhibit the activity of hydroxymethylglutaryl-coenzyme A reductase, are applied worldwide as lipid-lowering agents for dyslipidemia. Previous studies have reported that statins exert anti-inflammatory and anti-fibrotic effects in animal models of chronic liver diseases [112]. Although two recent studies, based on retrospective cohort studies, have suggested that statins may be beneficial in decreasing steatosis and fibrosis, as well as for inhibiting disease progression in patients with NAFLD [113,114], prospective studies are needed to confirm their effects.
Aramchol is an inhibitor of stearoyl-coenzyme A desaturase 1 (SCD1), which converts saturated fatty acids to monounsaturated fatty acids. Inhibition of SCD1 decreases fatty acid synthesis, which in turn reduces liver fat with improved insulin resistance [115]. In a phase 2 trial (NCT01094158), administration of aramchol for three months significantly reduced liver fat content in NAFLD patients [116]. Meanwhile, a phase 3 trial evaluating the efficacy of aramchol in NASH patients with fibrosis, is ongoing.
Thyroid hormone receptor beta (THR-β), which is highly expressed in hepatocytes, regulates lipid metabolism in the liver [117]. VK2809 and resmetirom (MGL-3196) are THR-β agonists that can activate lipid metabolism leading to improvements in NASH [118]. Phase 2 trials for these drugs have reported a reduction in liver fat and low-density lipoprotein cholesterol [119,120]. Additionally, VK2809 is being investigated for its efficacy and safety in a phase 2b trial for NASH patients (NCT04173065), while resmetirom is being assessed in a phase 3 trial for NASH patients with F2–3 fibrosis (NCT03900429).
FGF19 is a hormone involved in the regulation of bile acid metabolism [121]. Previous studies have reported reduced concentrations of circulating FGF19 and elevated bile acid concentration in NAFLD patients [122,123], suggesting that FGF19 dysregulation might be involved in NASH progression. NGM282, an FGF19 analog, inhibits bile acid synthesis without FGF19-related hepatocarcinogenesis [124,125]. In mouse models of NASH, NGM282 exerts anti-steatotic, anti-inflammatory, and antifibrotic effects without promoting liver tumorigenesis [126]. A recent phase 2 trial (NCT02443116) has demonstrated that NGM282 reduces liver fat content, as well as markers of liver inflammation and fibrosis in NASH patients [127].
Alternative causes of liver fibrosis
The development of antiviral agents against HBV and HCV is the most successful strategy to prevent liver fibrosis progression. In a trial including 348 patients with chronic hepatitis B, antiviral therapy with tenofovir resulted in regression of liver fibrosis in 51% of the participants, including patients with cirrhosis [10]. Other reports have shown that long-term viral suppression with entecavir lead to histologic improvement of liver fibrosis [11,128,129]. Similarly, sustained virologic response (SVR) by antiviral therapy for chronic HCV infection is also associated with liver fibrosis regression. A study reported that 50–60% of cirrhosis patients who achieved SVR by interferon therapy experienced histological regression of liver fibrosis [12,130]. Patients who achieved SVR following treatment with direct-acting antiviral agents also experienced liver fibrosis regression [131,132].
Corticosteroids and immunosuppressive agents are the primary drugs used to treat autoimmune hepatitis. In fact, corticosteroids have been shown to improve liver fibrosis in two-thirds of patients with autoimmune hepatitis [133]. Meanwhile, immunosuppressive therapies are not effective for PBC. However, ursodeoxycholic acid, the essential drug for PBC, reportedly delays the progression of liver fibrosis in patients with early stage PBC [134].
CONCLUSIONS
Liver fibrosis, including cirrhosis, is believed to be potentially reversible. Hence, it is essential to improve liver fibrosis to treat the underlying liver disorder. Many anti-fibrotic drugs targeting hepatocytes, HSCs, and immune cells are being investigated in clinical trials. However, the results of many of these trials suggest that treatment with single agents is not sufficient to ameliorate advanced liver fibrosis. Therefore, combination therapies comprising drugs that act on different mechanisms should be further investigated, along with the development of anti-fibrotic drugs with novel mechanisms. In the near future, therapeutic agents for liver cirrhosis will progress toward clinical application, exploiting the reversibility of liver fibrosis as a primary strategy.
Notes
Authors’ contribution
N.O. and T.M. wrote the manuscript and prepared the figures and tables. N.O., T.M., M.S.M., H.F., M.E., and N.K. revised the manuscript.
Conflicts of Interest: The authors have no conflicts to disclose.
Acknowledgements
N.K. was supported by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS) through Grant No. J192640002 (2019–2022) and by the Japan Agency for Medical Research and Development (AMED, 2019–2022).
Abbreviations
ALD
alcoholic liver disease
ASK1
apoptosis signal-regulating kinase 1
CBP
cyclic AMP-response element binding protein-binding protein
CCL
C-C chemokine ligand
CCL2
C-C motif ligand
CCl4
carbon tetrachloride
CCR
C-C chemokine receptor
CVC
cenicriviroc
CYGB
cytoglobin
DAMPs
damaged-associated molecular patterns
ECM
extracellular matrix
FDA
Food and Drug Administration
FXR
farnesoid X receptor
HBV
hepatitis B virus
HCV
hepatitis C virus
HSCs
hepatic stellate cells
HSP47
heat shock protein 47
HVPG
hepatic venous pressure gradient
ILs
interleukins
LOXL2
lysyl oxidase-like 2
MMPs
matrix metalloproteinases
NAFLD
non-alcoholic fatty liver disease
NASH
nonalcoholic steatohepatitis
NF-κB
nuclear factor-kappa B
NLRP3
NLR family pyrin domain containing 3
NOX
NADPH oxidase
NTZ
nitazoxanide
OCA
obeticholic acid
PAMPs
pathogen-associated molecular patterns
PBC
primary biliary cholangitis
PFD
pirfenidone
PRRs
pattern recognition receptors
qHSCs
quiescent hepatic stellate cells
ROS
reactive oxygen species
SCD1
stearoyl-coenzyme A desaturase 1
SVR
sustained virologic response
THR-β
thyroid hormone receptor beta
TNF-α
tumor necrosis factor-alpha