


INTRODUCTION
Primary biliary cholangitis (PBC), formally named primary biliary cirrhosis until 2016, is a chronic cholestatic liver disease. The first patient presenting with symptoms resembling PBC was described in the literature in 1851 [1]. When the term “primary biliary cirrhosis” initially appeared in the title of an article published in 1949 by Dauphinee and Sinclair [2], most early descriptions of PBC involved patients at the cirrhotic stage, with jaundice, ascites, and variceal bleeding; therefore, the nomenclature “primary biliary cirrhosis” was correct at that time. However, the use of biochemical and immunological tests in clinical settings has enabled the diagnosis of PBC at earlier stages. Furthermore, the establishment of ursodeoxycholic acid (UDCA) as a first-line treatment drug remarkably reduced disease progression to cirrhosis. In fact, while half of patients diagnosed as PBC had one or more symptoms at diagnosis in the 1980’s, 75–80% of patients were asymptomatic without any complaint of symptom at diagnosis after 2000 in Japan. The serious gap between the disease manifestation and its misnomer became wider, and the term “cirrhosis” became not merely an inaccuracy but an active stigma for patients, motivated by the change of nomenclature of the disease, from cirrhosis to cholangitis [3-10]. The Asian Pacific Association for the Study of Liver (APASL) also officially approved this decision, and the new nomenclature “primary biliary cholangitis” is currently used in the official journal of the APASL [11].
EPIDEMIOLOGY
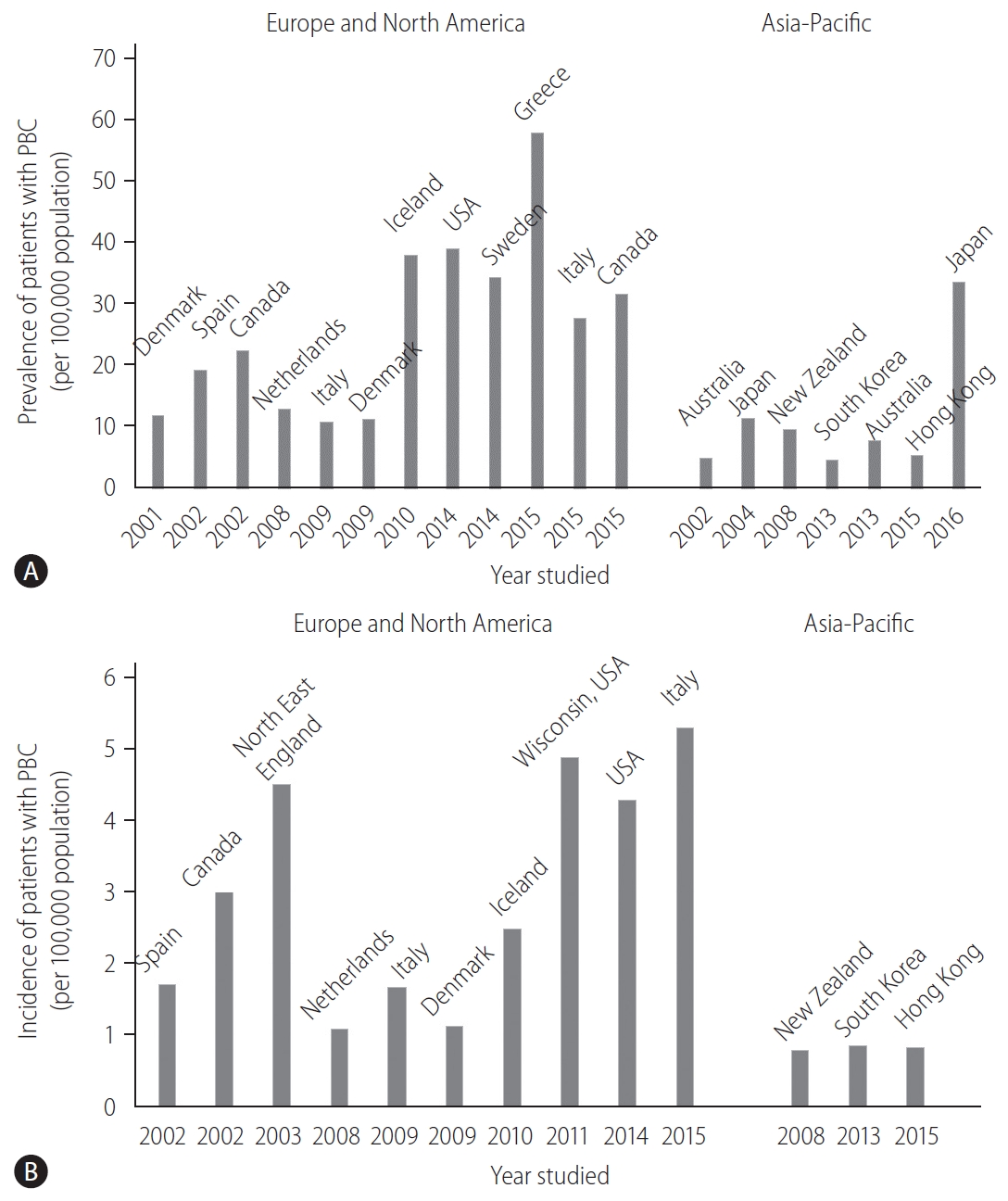
The prevalence and incidence of PBC vary considerably worldwide (Fig. 1). This discrepancy can be attributed to the true epidemiological difference between regions or study periods, to the variation in study designs for case finding or ascertainment, or difference in the diagnosis of PBC among physicians. Notably, awareness of PBC may still not be satisfactory in some Asian and African countries where epidemiological studies are scarce, and the sample size in some studies is very small. PBC is believed to be a rare disease in the Asia-Pacific region, and both the prevalence and incidence seemed to be lower in the Asia-Pacific region, as indicated by recent epidemiological studies in South Korea or Hong Kong (Fig. 1) [12,13]. In contrast, a 2016 study in Japan reported that the point prevalence of PBC was 33.8 per 100,000 in the Japanese population [14], which was comparable to that in European countries, the United States, and Canada. A recent review from South Korea defined a “rare disease” by either the prevalence (1,500–2,500 per 100,000 population) or patient number (<20,000) [15]. According to this definition, PBC is a rare disease, but interestingly, there is a substantial difference in the prevalence between Japan and South Korea or Hong Kong.
Other studies also indicated an increasing trend in the prevalence of PBC over time (Fig. 1A) [16]. Longitudinal studies in identical regions consistently showed an increase in the prevalence of PBC [14,17,18]. Since sequential studies demonstrated an increasing incidence of PBC (1.67 in 2009 and 5.31 in 2015, in Italy [19,20]) or a relatively stable incidence (2.6 in Sweden [18]), it is unclear whether a true increase or improved overall survival has contributed to the increasing prevalence of PBC.
One of the signatures of PBC is its female preponderance. Retrospective analysis indicated that the female:male ratio was 9:1 [21,22] in the 1990s and early 2000s, and this overt female predisposition has provided researchers a clue in clarifying the etiology of PBC. Notably, female predominance is still clearly observed today; however, it is less pronounced; the female:male ratio was less than 5:1 in most recent epidemiological studies and even 2.1:1 [19]. The reason for the relative increase in the number of male patients with PBC remains unclear, but a better recognition and a true increase in the incidence of PBC are likely responsible.
ETIOLOGY
Tolerance breakdown against autoantigens
PBC is a multifactorial and enigmatic disease; it remains unknown how and why PBC develops. Autoimmune attack targeted at biliary epithelial cells (BECs) through tolerance breakdown triggers disease onset.
The hallmark of PBC is anti-mitochondrial antibody (AMA), which are detected in 90–95% of patients with PBC [23,24], and the most disease-specific autoantibodies in human immunopathology. The high specificity of AMAs for PBC suggests that AMAs are not simple serological markers for diagnosis but are important in the immunopathology of PBC. AMAs recognize a family of enzymes located in the inner membrane of the mitochondria, named the 2-oxo-acid dehydrogenase complex (2-OADC), which mainly includes the pyruvate dehydrogenase complex E2 subunit (PDC-E2), the branched-chain 2-OADC E2 subunit (BCOADC-E2), the 2-oxoglutaric acid dehydrogenase complex E2 subunit (OGDC-E2), and dihydrolipoamide dehydrogenase-binding protein (E3BP) [25]. AMAs and autoreactive CD4+ and CD8+ T cell epitopes are confined within a shared peptide sequence of the inner lipoyl domain of human PDC-E2 [26].
BECs and hepatocytes of patients with PBC express large amounts of human leukocyte antigen (HLA) classes I and II molecules [27,28]. In patients with PBC, BECs acts as nonprofessional antigen-presenting cells, and the interplay of BECs and T cells may, to some extent, account for bile duct loss. Indeed, BECs expresses adhesion molecules, cytokines, and chemokines, and recruit mononuclear cells in the biliary tract of the liver. One example is fractalkine (CX3CL1), a chemokine with both chemoattractant and cell-adhesive functions [29]. T helper type 1-cytokine predominance and lipopolysaccharide in the microenvironment of injured bile ducts induce the upregulation of fractalkine expression in BECs, followed by the chemoattraction of mononuclear cells expressing its receptor (CX3CR1), including CD4+ and CD8+ T cells [30,31]. Serum fractalkine levels in PBC are high in patients with marked cholangitis activity (CA) at early stages, and they decrease in response to treatment [32].
PDC-E2 is a ubiquitous protein located in nearly all nucleated cells in the human body, and it remains unclear why autoreactive T cells specific for PDC-E2 elicit cytotoxicity only against BECs in the liver. In this regard, it should be noted that PBC recurs even after liver transplantation (LT), indicating that the immunopathological susceptibility of BECs in PBC is not major histocompatibility complex (MHC)-specific but a general feature shared with autologous BECs. The hypothesis to solve this enigma is that human intrahepatic BECs could maintain PDC-E2 immunologically intact within apoptotic blebs (apotopes) during apoptosis [33]. Interestingly, a unique triad consisting of BEC apotopes, macrophages from patients with PBC, and AMAs could lead to rigorous production of inflammatory cytokine production [34].
Genetic predisposition
Accumulating evidence also suggests that a combination of genetic predisposition and environmental triggering factors plays a crucial role in tolerance breakdown.
Genetic predisposition is believed to be a major contributing factor in the development of PBC [35].
A recent study in Iceland, which took advantage of the unique local genealogical database, demonstrated that the familial risk of PBC was present not only in first-degree relatives but also in second- and third-degree relatives of patients with PBC, with increased relative risk ratios (RRs) of 9.13 (95% confidence interval [CI], 4.17–16.76), 3.61 (1.48–8.92), and 2.59 (1.35–4.67), respectively [36]. Furthermore, the increased risk of PBC tended toward significance even in fourth- and fifth-degree relatives with RRs of 1.66 (1.00–3.02) and 1.42 (0.99–2.20), respectively. These findings clearly emphasize the importance of genetic risk in the pathogenesis of PBC.
Recent genome-wide association study (GWAS) analyses from North America, European countries, Japan, and China identified other HLA alleles that are strongly associated with susceptibility to PBC and revealed more than 40 non-HLA alleles contributing to PBC susceptibility (Table 1) [37-48]. Although risk alleles differ among studies and populations, they primarily belong to genes and pathways involved in antigen presentation and production of interleukin (IL)-12 (IRF5, SOCS1, TNFAIP3, NF-B, and IL-12A), activation of T cells, and interferon (IFN)-γ production (TNFSF15, IL12R, TYK2, STAT4, SOCS1, NF-κB, and TNFAIP3) as well as activation of B cells and production of immunoglobulins (POU2AF1, SPIB, PRKCB, IKZF3, and ARID3A). These immune pathways could be important in the pathogenesis of PBC.
Environmental triggers
As environmental triggers, large-scale case-control studies have consistently found an association between urinary tract infections and cigarette smoking with PBC [49-52]. Bacterial infection may have an impact on the etiology of PBC because PDC-E2 has a molecular mimic between human PDC-E2 and Escherichia coli (E. coli) PDC-E2, and thus, E. coli infection may trigger the breaking of immunological tolerance against human PDC-E2. Case-control studies also illustrate that xenobiotic modification of PDC-E2 with chemicals abundantly found in daily life, such as lipsticks, hair dyes, and nail polish, has a role in generating immunogenic neoantigens and breaking tolerance in PBC [51,53-57]. Finally, dysbiosis of the gut microbiota was found in patients with PBC, and interestingly, it was partially resolved with UDCA treatment [58].
Microbiota and bile acids
Early environmental exposures can influence microbiome development. Evidence for the role of the microbiome in the etiology of PBC includes the effect of diet and the higher prevalence of PBC in Western nations in the Northern hemisphere [59]. Diet is the primary external factor that can affect the microbiome, and changes in the microbiome can be affected by bile acids [60]. It is known that not all commensal bacteria are equally susceptible to bile acids [61], with E. coli and Helicobacter spp. being particularly resistant. Bile acids act as signaling molecules involved in glucose-lipid metabolism. The main bile acids in humans include cholic and chenodoxycholic acids and are synthesized in the liver [62]. Bile acids have several other effects that maintain healthy liver tissue, including the regulation of glucose tolerance and insulin sensitivity. Bile acids are also known to induce sterol regulatory element-binding protein-1c production, which regulates the biosynthesis of cholesterol, and also regulates the expression of lipogenesis genes such as glycerol-3-phosphate acyltransferase, acetyl-CoA carboxylase, and fatty acid synthase, thus offering protection against hyperlipidemia [63]. Besides their effects on liver homeostasis, bile acids may alter the microbiome and hepatic regeneration, and thereby play a role in the pathogenesis of PBC [64]. The chronic cholestasis seen in PBC that results from the microbiota affected by bile acids in turn can lead to aberrant expression of bile acid transporters and nuclear receptors, which can in turn cause liver damage, leading to a vicious cycle for chronic liver damage with bile acids as the pivotal determinant [65]. It is clear that the role of bile acids and bile salts in the pathogenesis of PBC is extremely complicated, and is influenced by a number of interacting mechanisms.
Studies on changing the microbiome to be more consistent with healthy controls can be achieved through the use of UDCA treatment, but the effects of generating a healthier “microbiome” on the development of autoimmune liver disease are still unclear. It is known that a healthy microbiome tends to be beneficial for products such as single chain fatty acids [66] and less proinflammatory cytokines such as TNFα and IFNγ [67].
DIAGNOSIS
A diagnosis of PBC is made when 2 or 3 following items are met: the 1) consistent elevation of cholestatic enzymes, 2) detection of AMA, and 3) typical liver histology [58,68,69]. Figure 2 shows a diagnostic flowchart. Because of the very high sensitivity and specificity of AMA in the diagnosis of PBC, detectable AMA and elevation of alkaline phosphatase (ALP) levels are adequate for the diagnosis of PBC, and liver biopsy is not mandatory in many cases. Nevertheless, since histopathological stage at baseline is an independent prognostic marker of survival, as demonstrated by a recent large-scale retrospective study [70], liver biopsy is recommended for predicting long-term outcome but is not necessary in making a diagnosis. A histological examination is required in atypical cases, including suspicious AMA-negative PBC and autoimmune hepatitis/PBC overlap.
In clinical settings, AMA is often determined by enzyme-linked immunosorbent assay (ELISA) or immunofluorescence. Traditionally, at least nine AMA subtypes have been identified, and four of them (M2, M4, M8, and M9) seemed to be associated with PBC [71]. Since anti-M2 antibody was directed to 2-OADC and the most specific antibody to PBC, the M2 subtype of AMA (AMA-M2) is currently used as AMA in daily clinical practice. In ELISA, a combination of three recombinant mitochondrial proteins (PDC-E2, BCOADC-E2, and OGDC-E2) was used as the antigen. The titer of AMAs is not associated with disease progression or the patient’s clinical course. AMAs are occasionally detected in less than 1% of healthy individuals with normal liver test results [72,73]. Individuals who are AMA-positive are at higher risk of developing PBC and require close follow-up, although the risk does not appear to be high, as previously believed. A large-scale cohort study in France demonstrated that the prevalence of AMA-positive patients without evidence of PBC was 16.1 per 100,000 population, and one in six patients with AMA positivity and a normal ALP level developed PBC within 5 years [74]. Conversely, a recent study from China demonstrated that more than 80% of patients with AMA without elevation of serum ALP levels also developed histological characteristics of PBC, suggesting the presence of undiagnosed PBC patients among those with normal ALP levels and AMA positivity [75]. It remains unclear whether these individuals will progress to advanced disease, as in typical PBC, and how they should be clinically treated.
Among several anti-nuclear antibodies, sp100 and gp210 are frequently found in the sera of patients with PBC, and, thus, aid in diagnosing patients with probable PBC but undetectable AMA positivity. A combination of three mitochondrial antigens, sp100, and gp210, (“PBC screen”) had a sensitivity of 83.8% and specificity of 94.7% for diagnosing PBC and was considered appropriate as the first-line screening test [76]. Molecular mimicry between mitochondrial antigens and sp100/gp210 was reported [77]. The detection of gp210 may be associated with progression of the disease in UDCA-treated patients [78], but this observation requires further validation.
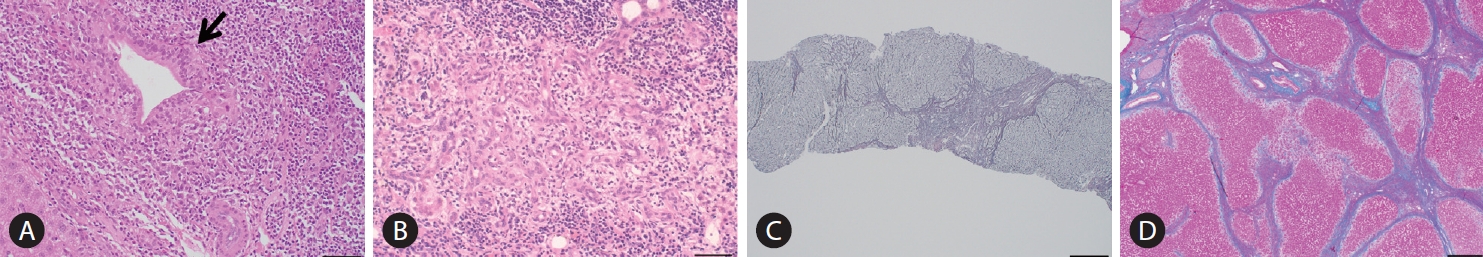
Histopathologically, the pathology of PBC is exclusively located in the intrahepatic small- or middle-sized bile ducts. A dense infiltration of mononuclear cells around the intrahepatic small bile ducts (interlobular bile ducts), coined as chronic non-suppurative destructive cholangitis (CNSDC), and granuloma formation are characteristic findings. Eventually, intrahepatic small bile ducts disappear from the liver, and chronic cholestatic features gradually develop. Hepatitis activity (HA) and chronic CA contribute to progressive hepatocellular damage and fibrosis, resulting in liver cirrhosis and hepatic failure.
The Scheuer’s [79] or Ludwig et al.’s [80] classifications has been used for a long time as the classification system in histological staging of PBC pathology. In Scheuer’s classification, florid duct lesions (CNSDC), ductular proliferation, scarring, and nodular cirrhosis are representative findings of stages 1, 2, 3, and 4, respectively (Fig. 3). However, as described in the original report by Scheuer, there is considerable overlap of findings between these stages; CNSDC can be observed even in the liver with nodular cirrhosis. Additionally, the pathology of PBC is not always distributed evenly in the liver; hence, sampling error can occur when determining the stages with these systems.
In order to overcome these limitations, Nakanuma et al. proposed a new histological staging and grading system for PBC (Tables 2-4) [81]. In Nakanuma’s classification, the scores for fibrosis, bile duct loss, and deposition of orcein-positive granules were used for staging, whereas CA and HA were used for grading. CA is determined by the presence of chronic cholangitis or CNSDC, and HA is defined by the presence of interface hepatitis or lobular hepatitis. Overall survival was stratified better with Nakanuma’s classification than with the classic system [82].
TREATMENT
UDCA
Since the first report demonstrating its efficacy for PBC [83], UDCA has dramatically altered the natural course of PBC and has been approved as a first-line therapy for PBC worldwide [58,69,84]. UDCA is used at a dose of 13–15 mg/kg/day, and is recommended for all patients with PBC with elevated liver biochemistry levels.
UDCA improves serum biochemical abnormalities, delays the histological progression and development of varices, and prolongs LT-free survival. Interestingly, a recent retrospective study on a large cohort demonstrated that the LT-free survival of UDCA-treated PBC patients was significantly improved compared to those that received no treatment and also in a population with incomplete biochemical response to UDCA [85].
Approximately 20–30% of patients with PBC exhibited incomplete biochemical responses to UDCA. The outcomes of these patients were significantly worse than those with complete responses to UDCA [85]. It is strongly recommended that patients with incomplete responses to UDCA commence second-line treatment in addition to UDCA. For this purpose, various criteria employing combinations of biochemical markers at 1 year after commencement of UDCA treatment have been proposed (Table 5) [86-95]. Therefore, the “UDCA response score,” which predicts treatment response before starting UDCA treatment with baseline clinical variables, has been proposed [96]. Very recently, the validity of the UDCA response score was demonstrated in a Japanere cohort [97].
Obeticholic acid (OCA)
OCA is a selective ligand of the farnesoid X receptor (FXR). In an international cohort, prospective, randomized, placebo-controlled trial (POISE trial), 217 patients with PBC who showed an incomplete response (serum ALP level >1.67×upper limit of normal [ULN]) or an abnormal total bilirubin level (<2×ULN), or were intolerant to UDCA were enrolled and received 5–10 mg of OCA, 10 mg of OCA, and a placebo for 1 year. The primary end point was an ALP level <1.67×ULN with >15% reduction from the baseline and normal bilirubin levels. Among those patients, 46–47% achieved the primary end point [98]. Consequently, OCA received accelerated approval from the United States Food and Drug Administration (FDA) approval on May 27, 2016.
Although OCA has become the long-awaited second-line drug officially approved for PBC, it is still unsatisfactory for several reasons. First, the response rate to OCA was at most 50%, which means that up to 50% of the patients did not respond to OCA. Second, pruritus, a symptom frequently experienced by patients with PBC, appeared as an adverse effect in 56–68% of those treated with OCA. Third, the appropriate treatment duration for OCA should be continued for patients refractory to UDCA; thus, OCA may be prescribed lifelong along with UDCA. Based on the high cost of OCA ($69,350 per year), its never-ending prescription places substantial economic burdens on patients and societies; thus, it is not cost-effective [99]. Finally, it has not yet been confirmed whether the primary end points (ALP level <1.67×ULN with >15% reduction from the baseline and normal bilirubin level) are associated with improvement of long-term outcomes. Therefore, follow-up studies of the POISE trial are required by the FDA; a phase 3 study is currently ongoing (COBALT, NCT02308111), and the interim results at 3-year treatment suggested biochemical efficacy and safety [100] as well as histological improvement [101] of OCA. Regardless, the safety concerns of OCA should not be underestimated as the FDA released a warning in September 2017 stating that the use of OCA in PBC patients with decompensated cirrhosis (Child-Pugh-Turcotte class B and C) was associated with clinical worsening or even death.
Fibrates
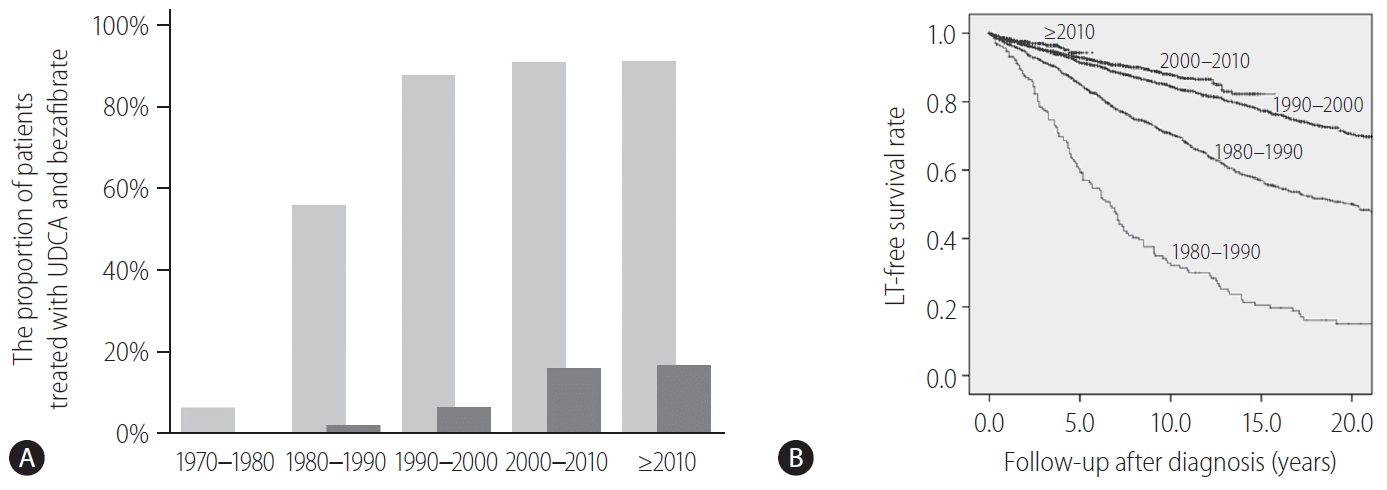
Fibrates are peroxisome proliferator-activated receptor-α and pregnane X receptor agonists and result in the reduction of de novo bile acid synthesis and upregulation of bile acid transporters [102]. Fibrates (fenofibrate and bezafibrate) were originally indicated for dyslipidemia and are used to decrease serum cholesterol and triglycerides. Bezafibrate was first reported as biologically effective for patients with PBC who were refractory to UDCA in 1999 [103]. A prospective, randomized, placebo-controlled study of bezafibrate in PBC patients with incomplete responses to UDCA from France demonstrated that an add-on of bezafibrate to UDCA for 2 years significantly improved liver biochemistry levels and liver stiffness [104]. In another study, additional evidence from Japan showed that the observed LT-free survival of patients treated with combination therapy of UDCA and bezafibrate was significantly superior to the expected LT-free survival of those treated with UDCA monotherapy according to the GLOBE and UK-PBC scores [105]. In Japan, bezafibrate has been used as a de facto second line treatment for patients who exhibited an incomplete response to UDCA. The proportion of patients treated with UDCA and bezafibrate and the LT-free survival stratified by the diagnosis year are shown in Figure 4. Until 1990, only 56% and 2% of patients were treated with UDCA and bezafibrate, respectively, and the LT-free 5-year survival rate of patients with PBC was 59%. By contrast, 91% and 16–17% of patients were treated with UDCA and bezafibrate after 2000, and the LT-free 5-year survival rate was significanltly improved to 93–94% (P<0.001). Bezafibrate may also improve pruritus of PBC [106]. A prospective clinical trial on this topic is ongoing (FITCH trial, NCT02701166).
Fenofibrate was reported to decrease serum ALP levels in studies from Japan and China [107,108], whereas the adjunct use of fenofibrate with UDCA showed no association with decreased serum ALP levels in a United Kingdom study [109]. Participants were recruited for a prospective randomized study in China (clinical trial ID: NCT02965911). However, these two prospective clinical trials showed notable improvements in liver enzyme levels at 12 or 24 months as the primary end point. Even after these trials were terminated, it is still unknown whether long-term outcomes are improved with additional treatment with fibrates, so follow-up studies of these trials are needed.
LT
Despite improvements in medical treatment for PBC, LT is the only treatment option for patients with decompensating events or intolerable pruritus. A recent study utilizing the European Liver Transplant Registry demonstrated a significant decrease in LT in PBC over the last 30 years after the introduction of UDCA in clinical settings [110]. The proportion of LT for PBC decreased from 20% of all LT cases in 1986 to 4% in 2015 (P<0.001). The absolute number of transplants was the highest in 1994 (n=279), which decreased to an average of 200 in the last decade. This decrease is striking in contrast to the substantial increase in the prevalence of PBC at the same time [16].
While the long-term outcome after LT for PBC is excellent overall [111-113], recurrence of PBC after LT is not uncommon. The reported incidence of recurrent PBC differs widely between 11% and 42% [114-129]. Although several studies have reported risk factors associated with recurrence of PBC, most consistently demonstrated that the use of tacrolimus is associated with an increased risk of recurrence [115,116,123,126-128]. For example, a recent study of 785 patients with PBC from North America and Europe who underwent LT between February 1983 and June 2016 indicated that tacrolimus was linked to the recurrence of PBC. Although the use of cyclosporine was protective, the 5-year probabilities of recurrence of PBC were reported to be 28% and 11% in patients receiving tacrolimus and cyclosporine, respectively (P<0.001) [125]. On the other hand, the role of tacrolimus as an increasing agent of recurrent PBC does not seem to be the case in other ethnicities. Recently, two cohort studies from Japan demonstrated that the increased frequency of recurrent PBC is associated with initial treatment with cyclosporine after LT [117,122].
Although it is believed that recurrence of PBC does not have a significant impact on long-term outcomes, such as overall survival [123,130], a recent study of 785 PBC patients from 13 centers in North America and Europe who received LT with a median follow-up of 6.9 years (interquartile range, 6.1–7.9) reported opposite and unexpected results that disease recurrence was found in 240 patients (31%). Importantly, graft and patient survival rates were significantly impaired in those with recurrent PBC (P=0.004 and 0.001, respectively) [125]. It is imperative to determine whether recurrent PBC has a clinically significant impact on patient and graft survival. Furthermore, since UDCA treatment seems to be effective in improving markers of cholestasis, it does not effectively reduce the frequency and risk of recurrence of PBC after LT [130]. Therefore, effective strategies to halt the recurrence of PBC are urgently needed.
Treatment flowchart
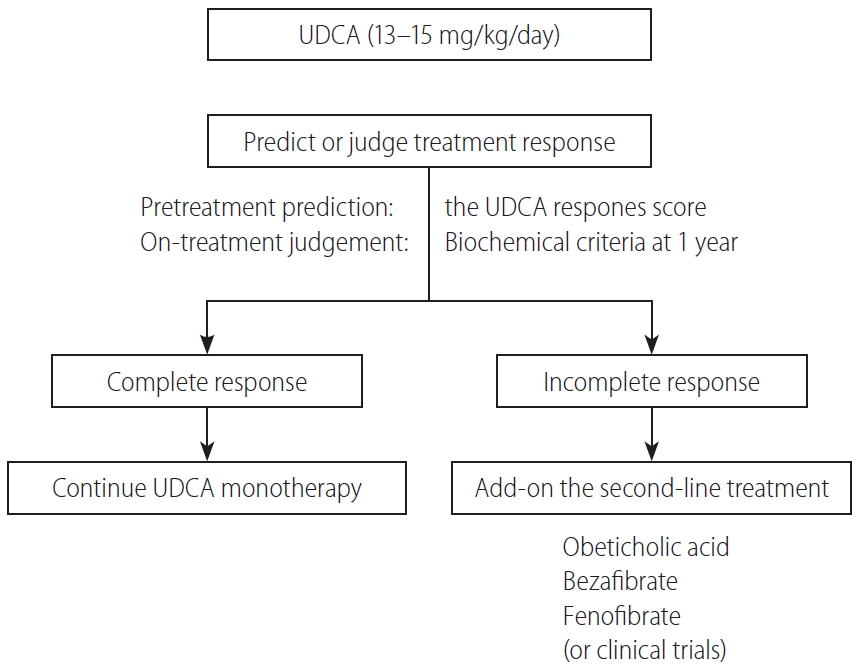
In Figure 5, a proposed treatment flowchart for patients with PBC is shown. UDCA treatment (13–15 mg/kg/day) is recommended for all patients diagnosed with PBC, with elevated cholestatic liver enzymes. Treatment response should be judged at 1 year of UDCA treatment with the biochemical criteria shown in Table 5. Since most newly diagnosed PBC patients are at an early stage in daily clinical practice, the Paris II criteria designed for early stage patients may be suitable and easy to use. If the treatment response is complete, UDCA monotherapy should be continued and no additional treatment is required. In patients with incomplete response, add-on of the second-line treatment is definitely necessary; otherwise, the long-term outcome would deteriorate. The evidence-based choice of second-line medication is very difficult because no drug has been demonstrated to improve long-term outcomes. In Japan, a de facto use of bezafibrate as a second-line treatment appears to contribute to the improvement of long-term outcome, and it is expected to demonstrate in a large scale cohort a significant association of a combination of UDCA and bezafibrate with reduced mortality or need for LT.
Alternatively, response to UDCA can be predicted before commencement of UDCA therapy using the UDCA Response Score, and initiation of treatment with a combination of UDCA and bezafibrate would be a choice in cases with prediction of poor response.
MANAGEMENT OF SYMPTOMS, EXTRAHEPATIC MANIFESTATIONS, AND HEPATOCELLULAR CARCINOMA (HCC)
Patients with PBC frequently present with numerous symptoms. The most dominant clinical symptoms in the early stage of PBC are fatigue and pruritus, followed by jaundice. Since these symptoms can significantly deteriorate the quality of life of patients with PBC, it is strongly recommended to carefully monitor these symptoms with objective and reproducible measures such as the PBC-40 questionnaire [131]. In Table 6, symptoms in PBC and corresponding recommended or emerging treatment options are summarized. In addition, along with an improvement in the long-term outcome of patients with PBC, the development of HCC is not a rare event.
Fatigue
Fatigue is the most common and debilitating symptom in PBC, experienced by approximately 50% of patients (ranging from 20% to 80% depending on each study) [132-134]. Although it is difficult to define the cutoff clearly according to the presence of fatigue, it has been repeatedly shown that fatigue has a great impact on the impairment of the quality of life of patients with PBC [133,135]. Fatigue is not associated with disease severity and staging, but may be related to age at onset and gender [136].
The cause of fatigue remains unknown but seems to be complex in origin, probably multifactorial in most patients and associated with depression, autonomic dysfunction, and sleep disturbance [135]. Recent studies with magnetic resonance imaging revealed that neuroimaging changes in the brain can be detected even in the early stage of PBC [137]. Fatigue cannot be treated with UDCA, and a recent systemic review failed to define any established treatment for fatigue in PBC [138]. Fatigue may be improved by LT, but it can also persist in a substantial portion of patients even after LT, making the role of LT as a therapeutic option for severe fatigue questionable [139]. Modafinil, which is officially approved by the FDA for wakefulness disorders, has been used, but a recent randomized, placebo-controlled clinical trial failed to indicate beneficial effects of this drug in reducing fatigue in patients with PBC [140].
Pruritus
Pruritus is another common symptom in PBC, affecting 20–80% of patients. Pruritus can occur locally or diffusely, and its presence and severity change throughout the clinical course of PBC. It tends to become more pronounced with the progression of PBC but can be present even in the very early stage. Pruritus can be highly bothersome and intolerable to patients, such as causing sleep disturbance, and is an important indication for LT. Similar to fatigue, the severity of pruritus is objectively assessable with the PBC-40 questionnaire [131]. The cause of pruritus remains unknown, although several substances are hypothesized to be related to pruritus in cholestatic liver diseases [141]. Most notably, lysophosphatidic acid (LPA) may be a potential candidate for initiating pruritus [142], and the activity of serum autotaxin, which converts lysophosphatidylcholine into LPA, and is related to the severity of pruritus and responds to therapeutic interventions [143,144]. Hence, LPA-autotaxin is an important candidate as a therapeutic target, yet clinically unavailable.
Although several lines of treatment options are recommended, severe pruritus is frequently intractable. In daily clinical practice, anion-exchange resins such as cholestyramine, rifampsin, and opiate antagonists are used [145]. Anion-exchange resins are recommended as the first-line therapy in the American Association for the Study of Liver Diseases and European Association for the Study of the Liver guidelines, despite its limited efficacy [68,146]. Cholestyramine is prescribed as 4 g per dose to a maximum of 16 g/day, and it is important to be administered 1 hour after or 4 hours before other medications (especially UDCA) to avoid inhibiting their absorption [68]. Rifampicin is a pregnane X receptor agonist that has been used for pruritus at 150–300 mg twice daily. Opiate antangonists interefere with increased endogenous opioid levels in patients with cholestatic pruritus [145]. While m-opioid receptor antagonists, such as naloxone or naltrexone, have been used, nalfurafine hydrochloride, a selective κ-opioid receptor agonist, is currently used in Japan for cholestatic pruritus and exhibits substantial efficacy [147]. As an emerging novel therapy, an ileal bile acid transporter (IBAT) inhibitor compound (linerixibat) that inhibits reabsorption of bile acids in the ileum effectively decreased pruritus in patients with PBC in a phase 2a study [148]. A global phase 2b study (clinical trial ID: NCT02966834) investigating the efficacy of linerixibat is almost complete. A phase 2 trial of another IBAT inhibitor, maralixibat, failed to demonstrate a significant anti-pruritic effect against placebo, presumably because of a significant placebo effect [149]. As described earlier, bezafibrate is now being investigated in a clinical trial for the treatment of cholestatic pruritus (clinical trial ID: NCT02701166) [106].
Sicca syndrome
The sicca complex is frequently present in patients with PBC, manifesting as dry eyes and/or dry mouth. External glands, including the lachrymal or salivary glands, are also affected in PBC. A current retrospective study revealed the prevalence of Sjögren’s syndrome in up to 56% of patients with PBC [150]; however, the sicca complex affects patients with PBC who do not meet the criteria of Sjögren’s syndrome. Patients with sicca syndrome may experience many symptoms, including burning, itching, irritated eyes, blepharitis, dysphagia, stomatitis, dental caries, and dry cough, resulting in severe impairment of their quality of life. Early recognition of sicca symptoms and consultations with ophthalmologists or dentists are suggested. Artifical tears and saliva are often helpful, and pilocarpine or cevimeline may alleviate the symptoms in refractory cases.
Osteopenia and osteoporosis
Osteopenic bone disease, including osteopenia and osteoporosis, is a common disorder in PBC that mainly affects middle-aged women and is associated with an increased risk for fragility fractures. The decrease in bone mineral density found in PBC is multifactorial. Chronic cholestasis leads to malabsorption and deficiency of vitamin D, which is essential for bone metabolism. Other factors associated with bone diseases include age, sex, low body mass index, history of fragility fracture, and advanced stage of PBC [151,152]. Intervention with bisphosphonates for patients with osteoporosis and those with a history of fragility fracture is safe and improves bone mineral density [153]; nonetheless, it remains unclear whether bisphosphonate use is associated with a decrease in fragility fractures. Recently, denosumab, a fully human monoclonal antibody against the receptor activator of nuclear factor-kappaB ligand (RANKL), was demonstrated to be effective for treating osteoporosis in patients with PBC [154]. Since RANK-RANKL signaling might be implicated in the pathogenesis of PBC [155], treatment with denosumab could be used to target both osteoporosis and PBC.
Hyperlipidemia and metabolic syndrome
Chronic cholestasis is a main feature of PBC, and consequently, hyperlipidemia is common and affects up to 80% of patients [156]. Several prospective studies have indicated that an increase in serum lipid levels is not associated with a higher risk for cardiovascular diseases related to atherosclerosis, and treatment for hyperlipidemia per se is not necessary. Notably, these studies were conducted in the 1990s when metabolic syndrome was relatively rare in patients with PBC. A recent study from Italy demonstrated that cardiovascular events developed more frequently in patients with metabolic syndrome [157], indicating the importance of treatment intervention for patients with hyperlipidemia if metabolic syndrome exists.
HCC
HCC is occasionally encountered in patients with PBC. The reported incidences and risk factors for developing HCC from several large-scale retrospective cohorts are summarized in Table 7. Surprisingly, the incidence rates (cases per 1,000 patient-years) of HCC in all patients with PBC are similar across different regions: 3.6 in Barcelona, Spain; 3.7 in Padova, Italy [158]; 3.6 in a nationwide study in Japan [159]; and 3.4 in an international cohort [160]. The incidence rate was exceptionally high (6.6) in a cohort from Beijing, China [161], presumably because of the high rate of individuals with previous hepatitis B virus (HBV) infection. A history of HBV infection was identified as an independent risk factor for HCC in this study. The incidence rate was higher in men than in women. In those studies, male sex and advanced histological stage independently contributed to the development of HCC [158-161]. Treatment response was included among possible risk factors only in the international cohort study, and biochemical non-response at 1 year of UDCA treatment (Paris-I not fulfilled) significantly increased the future risk of HCC (adjusted hazard ratio, 3.44; 95% CI, 1.65–7.14; P<0.0001) [160]. Taken together, close monitoring of HCC is strongly recommended for high-risk patients with PBC, such as male patients, those with advanced-stage disease, and non-responders to UDCA. The mean survival of patients who developed HCC was 36 months after diagnosis, and another cohort indicated 5- and 10-year survival rates of 49.5% and 31.7%, respectively [162].
FUTURE PERSPECTIVE
Although the long-term outcome of PBC has been significantly improved, there remain several unmet needs in PBC. UDCA and other second-line treatments have little efficacy for patients with advanced disease, for whom LT is the only therapeutic option. No “cure” is achieved with current treatment, and patients are required to take medications for life long. A variety of symptoms, including fatigue and pruritus, are very difficult to manage and easily reduce the health-related quality of life.
To overcome these unmet needs in this field, rigorous efforts should be directed at improving our understanding of the environmental etiology and genetic basis of PBC, molecular mechanisms of disease progression, and gender bias to identify critical pathways for therapeutic interventions. Relevant animal models that recapitulate human PBC should be established for preclinical studies with designer drugs guided by this new knowledge. Our goal is achieving the “cure” for PBC.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print





