Oxidative stress and glutamate excretion in alcoholic steatosis: Metabolic synapse between hepatocyte and stellate cell
Article information

Abstract
Chronic alcohol consumption induces the development of alcoholic steatosis in the liver, which is one of the most widespread liver diseases worldwide. During general alcohol metabolism, hepatocytes generate mitochondria- and cytochrome P450 2E1 (CYP2E1)-mediated reactive oxygen species (ROS) whose accumulation elicits activation of the hepatic anti-oxidant system, including glutathione (GSH). However, chronic alcohol consumption decreases GSH generation through cysteine deficiency by suppressing the methionine cycle and trans-sulfuration system, whereas it turns on an alternative defense pathway, such as the xCT transporter, to compensate for GSH shortage. The xCT transporter mediates the uptake of cystine coupled to the efflux of glutamate, leading to an increase in blood glutamate. In response to the elevated glutamate in the liver, the expression of metabotropic glutamate receptor 5 (mGluR5) is up-regulated in hepatic stellate cells (HSCs) along with enhanced production of 2-arachidonoylglycerol, which in turn stimulates cannabinoid receptor 1 (CB1R) on neighboring hepatocytes to increase de novo lipogenesis. On the other hand, blockade of mGluR5 and CB1R attenuates alcoholic steatosis. Interestingly, although the increased expression of CYP2E1-mediated xCT and ROS generation are mainly observed at the perivenous area (zone 3), fat accumulation is mostly detected at hepatic zone 2. To resolve this discrepancy, this review summarizes recent advances on glutamate/mGluR5-derived alcoholic steatosis and zone-dependently different responses to alcohol intake. In addition, the bidirectional loop pathway and its unique metabolic synapse between hepatocytes and HSCs are discussed.
INTRODUCTION
In recent years, the tremendous increase in chronic alcohol consumption has become a global health threat. In 2018, the World Health Organization reported that 3 million people per year die as a result of the abuse of alcohol [1-3]. Chronic alcohol intake leads to a broad spectrum of alcoholic liver disease (ALD) such as simple steatosis, steatohepatitis, hepatitis, fibrosis/cirrhosis, and hepatocellular carcinoma [2,4]. Most individuals consuming over 40 g of alcohol per day are known to develop alcoholic steatosis, or alcoholic fatty liver, but only a subgroup of them progresses to more advanced ALD [3]. Several plausible factors such as genetic, epigenetic, and non-genetic issues might contribute to the individual differences in the progression of ALD [1,3]. Although the underlying mechanisms of ALD have been explored for decades, precise treatments for each type of ALD have not been developed yet. Nevertheless, intervention of ALD at the earliest stage is very important since it is difficult to reverse the progression at the late stages of liver diseases, which include liver fibrosis and tumors.
The pathogenesis in the early stages of ALD, including alcoholic steatosis, also varies with oxidative stress, alcohol metabolite-mediated toxicity, and activation of diverse inflammatory signals. In this review, we briefly describe the underlying mechanisms of oxidative stress-mediated glutamate secretion and endocannabinoid production in alcoholic steatosis and suggest a novel metabolic synapse between hepatic stellate cells (HSCs) and hepatocytes. In addition, we address different responses of alcohol metabolism that are specific to hepatic zones (Fig. 1).
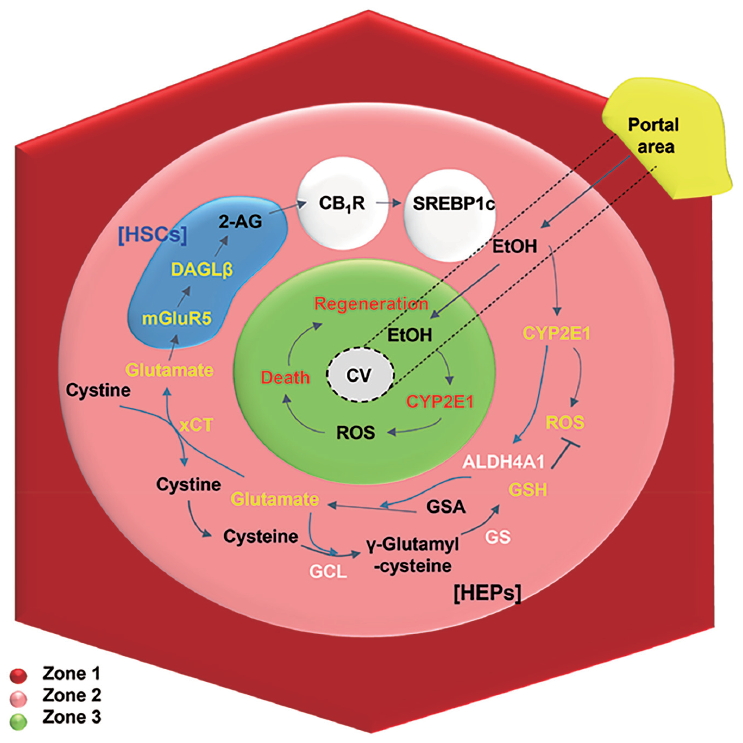
Hepatic zonal-dependent fat accumulation in the early stage of ALD. On chronic ethanol consumption, ethanol is mainly metabolized in perivenous hepatocytes with high expression of CYP2E1, inducing metabolic stress-mediated death and regeneration of hepatocytes at zone 3. From zone 3 to 2, CYP2E1-mediated ROS can be eliminated by GSH generation through xCT-mediated uptake of cystine, while excreted glutamate stimulates mGluR5 of HSCs to produce 2-AG, followed by up-regulated lipogenesis of hepatocytes through CB1R-mediated SREBP1c expression. HSC, hepatic stellate cell; 2-AG, 2-arachidonoylglycerol; CB1R, cannabinoid receptor 1; SREBP1c, sterol regulatory element-binding protein 1; DAGL, diacylglycerol lipase; mGluR5, metabotropic glutamate receptor 5; CV, central vein; CYP2E1, cytochrome P450 2E1; ROS, reactive oxygen species; GSA, glutamic-γ-semialdehyde; GSH, glutathione; GS, glutathione synthase; GCL, glutamate cysteine ligase; HEPs, hepatocytes; ALD, alcoholic liver disease.
LIVER ZONATION AND ETHANOL METABOLISM
The liver has numerous functions including production and breakdown of diverse proteins, detoxification against xenobiotics, and metabolic homeostasis of glucose, lipids, and cholesterols. The composition of repeating hexagonal units termed lobules is a specific feature of the liver. Blood supply from portal triad at the corner of the lobules towards central veins makes gradients of oxygen, nutrients, xenobiotics, and hormones along the sinusoid [5-7]. This graded environment assigns heterogeneity in gene expression to hepatocytes, which leads them to position at different lobular layers, a phenomenon called liver or hepatic zonation (Fig. 2A). Hepatocytes are layered from the portal to central vein, and are divided into three zones: zone 1 (periportal), zone 3 (perivenous), and zone 2, which refers to the area between zones 1 and 3. Recent single-cell RNA sequencing analysis of mouse and human livers has enabled the disclosure of heterogenic characteristics of liver zonation in precise detail [6,7]. As for cytochrome P450 2E1 (CYP2E1), its mild expression is normally detected in hepatocytes near the perivenous area (zone 3), gradually increasing towards the midzonal area (zone 2) following chronic alcohol consumption [2]. In contrast, carbamoyl phosphate synthetase I is mainly expressed in hepatocytes at the periportal area (zone 1) [5]. This was proven by the zonation profiles from a single-cell RNA sequencing analysis that revealed high expression of CYP2E1 in 3 to 5 layers of hepatocytes near zone 3 [6,7].
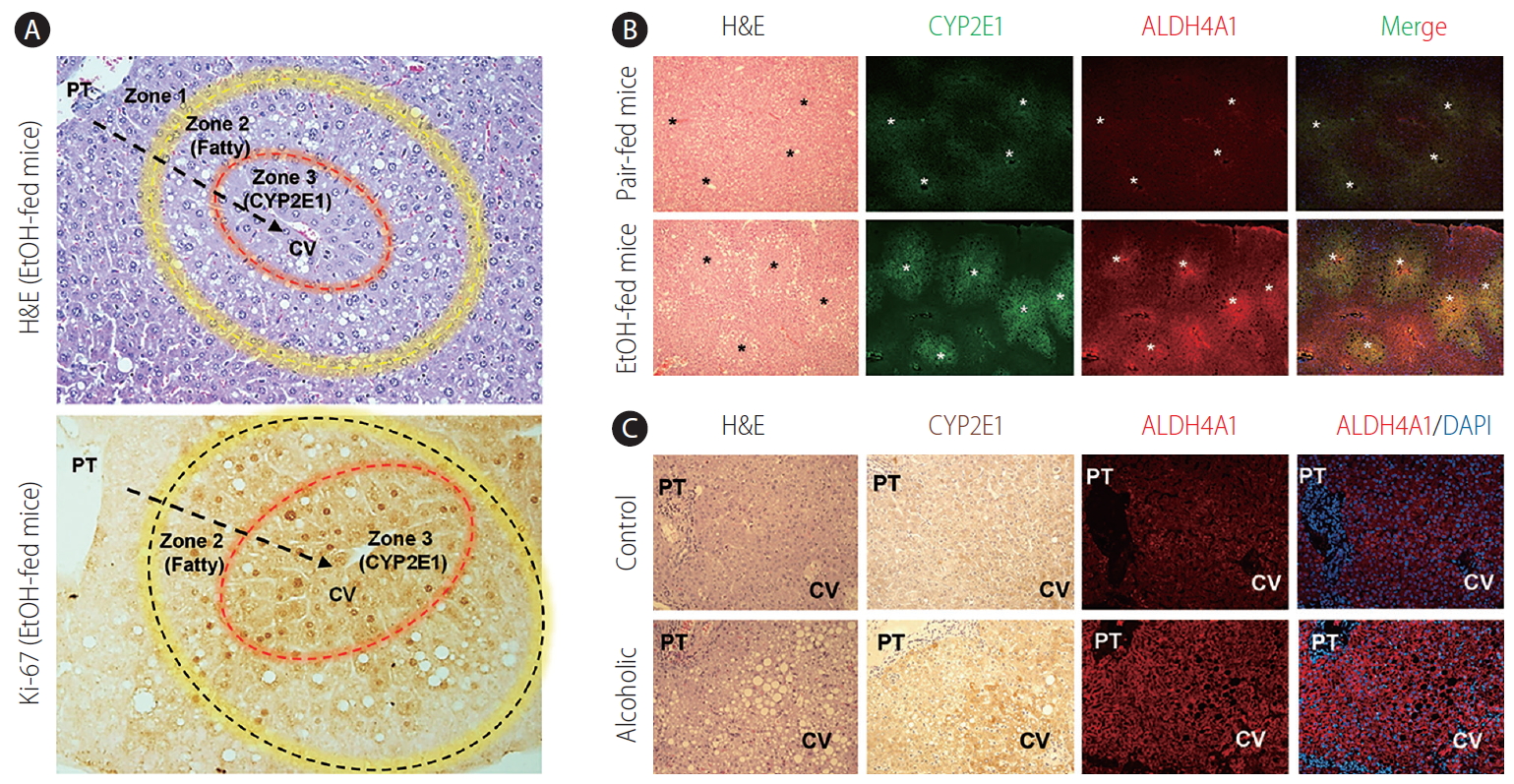
Hepatic zonal-dependent expression of CYP2E1 and ALDH4A1 in mice and patients with ALD. (A) After chronic alcohol consumption (4.5% of EtOH containing diet feeding for 8 weeks), fat accumulation is mainly observed at zone 2, whereas hepatic proliferation (Ki-67-positive) is mostly detected in zone 3 in stained liver sections of EtOH-fed mice. Arrows and asterisks indicate direction of blood flow and central veins, respectively. (B, C) Increased expression of CYP2E1 and ALDH4A1 is typically perceived around central veins of liver sections in EtOH-fed mice and alcoholic patients compared to their corresponding controls. Original magnification, ×200. H&E, Hematoxylin & Eosin; PT, portal area; CYP2E1, cytochrome P450 2E1; CV, central vein.
Although several studies have demonstrated that alcohol can be metabolized in brain, stomach, and adipose tissues, the major organ for alcohol metabolism is the liver. In fact, among various types of hepatic cells, hepatocytes mainly metabolize alcohol [8,9]. Alcohol metabolism starts with oxidation of the absorbed alcohol to acetaldehyde by alcohol dehydrogenase (ADH) in the cytoplasm, owing to the high affinity of ADH against alcohol (Km=0.2–2.0 mM), which is further metabolized by acetaldehyde dehydrogenase to acetate in the mitochondria of hepatocytes [3]. In chronic alcohol consumption, CYP2E1 plays critical roles in alcohol metabolism despite its lower affinity against alcohol (Km=8.0–10.0 mM) compared to ADH, and this is due to highly increased expression of CYP2E1 proteins (Fig. 2B). CYP2E1, which resides in microsomes, the endoplasmic reticulum (ER) or mitochondria in hepatocytes, metabolizes alcohol in the presence of nicotinamide adenine dinucleotide phosphate and oxygen, leading to the generation of reactive oxygen species (ROS) [3]. Once alcohol is oxidized to acetaldehyde via the oxidative pathways, it is rapidly further metabolized into acetate by aldehyde dehydrogenase 2 (ALDH2) in the mitochondria. ALDH2 generates nicotinamide adenine dinucleotide (NADH) as a byproduct, and oxidation of NADH by the mitochondrial respiratory chain produces ROS [10]. As a final product of alcohol metabolism, acetate is released to the circulation system.
The produced ROS mediates the damage of DNA, proteins, and lipids, and accounts for perivenous necrosis/apoptosis and regeneration of hepatocytes (Ki-67-positive cells at zone 3) (Fig. 2A). Interestingly, chronic alcohol consumption predominantly induces hepatic injury at zone 3, whereas fat accumulation is mainly observed, and is shown to start, in hepatocytes near zone 2 in livers of mice and patients with ALD (Fig. 2B, C) [11]. Thus, different pathological features depending on the zonation and the molecular mechanisms involved should be investigated clearly. In this review, we suggest a possible explanation for one of the unrevealed pathological mechanisms of fat accumulation at zone 2 following chronic alcohol consumption.
GLUTATHIONE (GSH) SYSTEM IN LIVER
The balance between alcohol-induced oxidative stress and recovery is tightly regulated in the context of liver homeostasis, in which CYP2E1-induced ROS contributes to the increased expression of multiple genes linked to anti-oxidative responses [3,10]. For example, it elevates the expression of transcription factor Nrf2, which leads to the upregulation of genes related to GSH synthesis [10]. Thus, GSH can alleviate the progression of ALD in heavy drinkers [3,12].
GSH synthesis pathway
In the cytosol of most cells, GSH, a cellular antioxidant defense system, is synthesized by two ATP-requiring enzymatic steps [13]. As the first step, glutamate cysteine ligase (GCL) synthesizes γ-glutamylcysteine from glutamate and cysteine, followed by GSH synthase (GS) producing GSH using γ-glutamylcysteine and glycine (Fig. 1). The intracellular concentration of glutamate in the liver is high enough for GSH synthesis while the cysteine concentration is similar with the Km value of GCL at a steady state, which means that cysteine acts as a rate-limiting substrate. In the liver, the methionine cycle and trans-sulfuration pathway provide cysteine for GSH synthesis.
Methionine cycle and trans-sulfuration pathway
Methionine is an essential amino acid that acts as a substrate for other amino acids such as taurine and cysteine. In the methionine cycle, methionine is converted to S-adenosylmethionine (SAMe) by methionine adenosyltransferase. By losing its methyl group by methyltransferase, SAMe becomes S-adenosylhomocysteine (SAH) and further hydrolysis of SAH by SAH hydrolase reversibly forms homocysteine [14]. Once homocysteine is formed, it can be remethylated to generate methionine or can be further converted into cysteine via the trans-sulfuration pathway [15]. The trans-sulfuration pathway is particularly activated in the liver to utilize methionine for GSH synthesis. In the trans-sulfuration pathway, cystathionine β synthase catalyzes homocysteine to form cystathionine, which is then cleaved by γ-cystathionase, releasing cysteine for GSH synthesis [15].
Dysregulation of GSH synthesis on alcohol consumption
Unlike in a healthy state, chronic alcohol consumption leads to an impaired methionine cycle and trans-sulfuration pathway, which causes accumulation of homocysteine in the ER and a shortage of cysteine, resulting in decreased GSH levels [16-18]. Consequently, the disturbed equilibrium between ROS and GSH leads to hepatic injury and inflammation. In fact, several studies in mice and patients with ALD have shown decreased hepatic GSH levels along with hyper-homocysteinemia, as well as increased lipogenic gene expression in hepatocytes [19-22]. Nevertheless, ROS-induced hepatic injury is not so severe in alcoholic steatosis, suggesting an alternative source of cysteine for GSH synthesis.
In the impaired methionine cycle and trans-sulfuration pathway, xCT transporter (encoded by Slc7a11) can compensate for the decreased cysteine levels through the exchange of cytosolic glutamate with extracellular cystine, where cystine is then converted into cysteine to form GSH [23]. In our recent study, we proposed a novel compensatory mechanism for the shortage of GSH and cysteine that involves robust induction of xCT and Nrf2 in hepatocytes of ethanol-fed mice [24]. Interestingly, xCT expression was shown to be co-localized with CYP2E1 in perivenous and midzonal hepatocytes in both mice and humans, suggesting a strong link between CYP2E1-mediated oxidative stress and xCT induction [24]. In regard to this, we also showed that xCT-mediated cystine uptake and glutamate excretion occur predominantly around these areas (zones 2 and 3). Taken together, these findings provide a plausible explanation for both hepatic necrosis (zone 3) and fat accumulation (zone 2), which are mainly due to gradient levels of ROS within hepatic lobules (Fig. 1). However, except for the exchange of cystine with glutamate, other roles of xCT transporter has not been elucidated clearly.
GLUTAMATE IN LIVER
Metabolism and role of glutamate
In addition to GSH synthesis, glutamate is involved in multiple metabolic pathways in the liver. Hepatocytes residing at periportal areas (zone 1) mainly convert glutamine to glutamate by glutaminase 2, in which the generated ammonia is then detoxified into urea, whereas perivenous hepatocytes at zone 3 express glutamine synthetase that conversely produces glutamine using glutamate and ammonia [25]. As for the generation of energy, the glutamate is converted into α-ketoglutarate by glutamate dehydrogenase, which further enters the tricarboxylic acid cycle to generate energy for cell growth and proliferation [26]. Glutamate is also involved in proline and ornithine metabolism, where glutamate is converted to pyrroline-5-carboxylate (P5C) through P5C synthase and further metabolized into proline or ornithine by P5C reductase or ornithine aminotransferase, respectively. Interestingly, P5C is conversely converted to glutamate by aldehyde dehydrogenase 4 family member A1 (ALDH4A1) [27].
According to our recent findings, chronic alcohol consumption increases ALDH4A1-mediated glutamate generation in perivenous hepatocytes in both mice and humans (Fig. 2B). However, the expression of other genes involved in glutamate generation such as Gls, Glul, Glud1, P5cr2, and Prodh remained unchanged, suggesting that glutamine may not be the source of alcohol-induced increase in glutamate in the liver [28]. Taken together, the findings indicate that the glutamate-driven amino acid metabolic pathway plays crucial roles in cell growth, protein metabolism, ammonia detoxification, and anti-oxidation, emphasizing that hepatic levels of glutamate/glutamine are important on the pathogenic conditions.
ENDOCANNABINOID IN LIVER
Endocannabinoid system
The endocannabinoid system consists of endogenous cannabinoids (endocannabinoids), cannabinoid receptors (CBR), and biosynthetic and degrading enzymes [29]. Endocannabinoids, including N-arachidonoylethanolamine (AEA), also known as anandamide, and 2-arachidonoylglycerol (2-AG), bind to cannabinoid receptor 1 (CB1R), which mediates effects of marijuana in the brain, as well as in peripheral organs including the liver. 2-AG is catalyzed mainly by diacylglycerol lipase (DAGL)-β in the liver and DAGLα in the brain, and degraded primarily by monoacylglycerol lipase [30,31]. AEA is produced from N-arachidonoyl-phosphatidylethanolamine by N-acyl-phosphatidylethanolamine-specific phospholipase D, and fatty acid amide hydrolase mediates degradation of AEA [32,33]. In the liver, CB1R activation contributes to various liver diseases, such as non-alcoholic fatty liver disease, alcoholic steatosis, inflammation, and fibrosis, whereas cannabinoid receptor 2 (CB2R) activation ameliorates the progression of liver disease by inhibiting activation of Kupffer cells [30,34-38]. Interestingly, both receptors are expressed in most hepatic cells including hepatocytes, HSCs, and immune cells [32].
Endocannabinoid in ALD
Alcohol stimulates 2-AG production in cerebellar granule neurons in rats [39] and HSCs in mice [30], which has been implicated in alcohol reward and dependence [40] and alcoholic steatosis [30], respectively. In addition, chronic alcohol consumption upregulates the expression of CB1R (encoded by Cnr1) in hepatocytes [30]. In fact, one of the key mechanisms mediating the development of alcoholic steatosis is the paracrine activation of CB1R in hepatocytes by HSC-derived 2-AG [30]. The stimulated CB1R drives de novo lipogenesis by up-regulating the expression of lipogenic genes such as Srebf1 and Fasn, while inhibition of CB1R attenuates alcoholic steatosis in the liver [30]. The molecular mechanism by which alcohol intake stimulates 2-AG production in HSCs has been recently elucidated and will be precisely addressed later in this paper. Another interesting study has demonstrated that CB1R activation induces the expression of CYP2E1 through estrogen-related receptor γ (ERRγ), a constitutively active transcriptional activator, on chronic alcohol consumption [41]. In this study, CYP2E1-mediated ROS generation was decreased in both CB1R-deficient and ERRγ inverse agonist-treated mice compared to control mice, suggesting a potential role of the endocannabinoid system in alcohol-induced hepatic liver injury and inflammation [41]. Hepatic endocannabinoid signaling is also associated with alcoholic liver fibrosis, with CB1R-deficient mice showing attenuated alcohol-induced inflammation and fibrosis, but CB2R-deficient mice presented the complete opposite phenotypes, which increased inflammation and fibrosis [38]. These results suggest that CBR have diverse roles in the development of ALDs and their inhibition might be a therapeutic approach for ALD.
GLUTAMATERGIC SIGNALING IN ENDOCANNABINOID PRODUCTION
Previously, we have demonstrated that chronic alcohol consumption increases the hepatic 2-AG production through DAGL-β in HSCs, and the released 2-AG induces CB1R-mediated de novo lipogenesis in hepatocytes [30]. However, the metabolic driver for the production of 2-AG in HSCs remained unidentified.
In the mammalian central nervous system (CNS), glutamate plays a role as a principal excitatory neurotransmitter, and glutamate released from presynaptic neurons binds to its receptors, including the metabotropic glutamate receptors (mGluRs), on postsynaptic neurons, which then activates downstream signaling pathways in the synapse [42]. There are three groups of mGluRs, and unlike the inhibitory group II and group III receptors, the group I receptors (mGluR1 and mGluR5) induce the synthesis of diacylglycerol by activating phospholipase C, which is further converted to 2-AG by DAGLα or DAGLβ [43]. In the CNS, endocannabinoids modulate the synaptic plasticity by activating presynaptic CB1R to inhibit glutamatergic (excitatory) transmission, following the release of endocannabinoids from post-synaptic neurons in response to metabotropic receptor activation [44]. There is a stunning analogy between endocannabinoid signaling in the CNS and the liver. Our previous studies suggest that a similar bidirectional signaling operates at a metabolic ‘synapse’ between hepatocytes (analogous to presynaptic neurons expressing CB1R) and HSCs (analogous to postsynaptic neurons generating 2-AG in response to mGluR5). Here, we provide multiple lines of evidence for the metabolic synapse [24,30].
Alcoholic oxidative stress-induced glutamate excretion triggers 2-AG production in HSCs through mGluR5. Upon alcoholic oxidative stress, cysteine deficiency and glutathione depletion occur and activate a compensatory mechanism through the up-regulation of xCT expression in hepatocytes, resulting in increased glutamate secretion. Glutamate then stimulates mGluR5 activation on neighboring HSCs to induce DAGLβ-dependent 2-AG production, and the released 2-AG activates CB1R on hepatocytes to induce de novo lipogenesis. Moreover, we demonstrated that xCT or mGluR5 inhibition by pharmacological blockade or genetic deletion alleviates alcoholic steatosis, whereas exposure of HSCs to mGluR5 agonists promotes DAGLβ expression and 2-AG production. Furthermore, patients with alcoholic steatosis showed elevated serum glutamate levels, increased expression of hepatic mGluR5 and xCT, and a positive correlation between mGluR5 and CB1R-mediated de novo lipogenesis. Hence, these components involved in the metabolic synapse offer potential as novel therapeutic targets for the intervention of alcoholic steatosis.
In terms of mGluR5 expression, the exact mechanism by which HSCs regulate and increase the expression of mGluR5 in response to alcohol consumption is still unclear. In the central nervous system, calmodulin stabilizes the surface expression of mGluR5 whereas protein kinase C (PKC) decreases the surface expression of mGluR5 [45]. In contrast, in the liver, acetaldehyde produced by alcohol metabolism stimulates PKC activation in HSCs to produce collagen fibers [46], suggesting a negative regulation of mGluR5 expression in HSCs by the activated PKC. In our study, however, chronic alcohol consumption induced a remarkable increase in mGluR5 expression in HSCs, suggesting that mGluR5 expression may be regulated by a different mechanism in HSCs. Thus, the exact molecular mechanism is yet to be elucidated and should be further investigated in the near future.
CONCLUSION
For the past two decades, the liver has been considered a metabolic or an immunological organ, which is why most investigations on the treatment of ALD have focused on the regulation of alcohol metabolism in hepatocytes or the hepatic immune system (e.g., Kupffer cells and toll-like receptors). Despite a great number of studies, there is still no specific treatment for ALD except abstinence. Lately, the importance of neurological pathways has emerged in the field of ALD and, in particular, the hepatic endocannabinoid system has drawn attention as a key mechanism of alcoholic steatosis development through paracrine activation of hepatic CB1R by HSCs-derived 2-AG, resulting in de novo lipogenesis in hepatocytes.
Our recent study revealed that chronic alcohol consumption increases the expression of xCT as a compensatory mechanism for the shortage of GSH in the perivenous hepatocytes by increasing cystine uptake and glutamate excretion through the xCT. The excreted glutamate binds to mGluR5 on neighboring HSCs to trigger the production of 2-AG, which then stimulates CB1R on hepatocytes. Having shown the importance of this bidirectional loop pathway between hepatocytes and HSCs, targeting this metabolic synapse could prevent the development of alcoholic steatosis. Therefore, further research on the glutamatergic signaling pathways in the liver should be carried out as these pathways may provide a potential therapeutic paradigm for the treatment of ALD.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grants funded by the Korea government (MEST) (2018R1A2A1A05077608), Korea Mouse Phenotyping Project (2014M3A9D5A01073556), and the KAIST Grand Challenge 30 Project (KC30), in 2019 (N11190207) funded Ministry of Science and ICT and KAIST, Korea.
Notes
Authors’ contributions
Hee-Hoon Kim entirely wrote the manuscript. Sung Eun Choi contributed to the editing of the manuscript and preparation of photos and images. Won-Il Jeong supervised the whole paper writing and edited the manuscript.
Conflicts of Interest
The authors have no conflicts to disclose.
Abbreviations
2-AG
2-arachidonoylglycerol
ADH
alcohol dehydrogenase
AEA
N-arachidonoylethanolamine
ALD
alcoholic liver disease
ALDH2
aldehyde dehydrogenase 2
ALDH4A1
aldehyde dehydrogenase 4 family member A1
CB1R
cannabinoid receptor 1
CB2R
cannabinoid receptor 2
CBR
cannabinoid receptors
CNS
central nervous system
CYP2E1
cytochrome P450 2E1
DAGL
diacylglycerol lipase
ER
endoplasmic reticulum
ERRγ
estrogen-related receptor γ
GCL
glutamate cysteine ligase
GS
glutathione synthase
GSH
glutathione
HSC
hepatic stellate cell
mGluRs
metabotropic glutamate receptors
NADH
nicotinamide adenine dinucleotide
P5C
pyrroline-5-carboxylate
PKC
protein kinase C
ROS
reactive oxygen species
SAH
S-adenosylhomocysteine
SAMe
S-adenosylmethionine