From intestinal dysbiosis to alcohol-associated liver disease
Article information

Abstract
Alcohol-associated intestinal dysbiosis and bacterial overgrowth can lead to a dysregulation of tryptophan metabolism and lower production of indoles. Several of these indole derivatives are aryl hydrocarbon receptor ligands that, in turn, are involved in antimicrobial defense via induction of interleukin-22 (IL-22). IL-22 increases the expression of intestinal regenerating islet-derived 3 (Reg3) lectins, which maintain low bacterial colonization of the inner mucus layer and reduce bacterial translocation to the liver. Chronic alcohol consumption is associated with reduced intestinal expression of Reg3β and Reg3γ, increased numbers of mucosa-associated bacteria and bacterial translocation. Translocated microbial products and viable bacteria reach the liver and activate the innate immune system. Release of inflammatory molecules promotes inflammation, contributes to hepatocyte death and results in a fibrotic response. This review summarizes the mechanisms by which chronic alcohol intake changes the gut microbiota and contributes to alcohol-associated liver disease by changing microbial-derived metabolites.
INTRODUCTION
Alcohol consumption is one of the major causes of chronic liver disease in Western countries. In the United States, according to the 2018 National Survey on Drug Use and Health, 14.4 million adults ages 18 and older (5.8% of this age group) had alcohol use disorder, including 9.2 million men (7.6% of men in this age group) and 5.3 million women (4.1% of women in this age group). In 2018, of the 83,517 liver disease deaths among individuals ages 12 and older, 47.8% involved alcohol [1].
The global burden of alcohol-associated liver disease is immense and comprises relatively mild and reversible alcohol-associated hepatic steatosis (fatty liver) to fibrosis and cirrhosis, and alcoholic hepatitis [2,3]. Besides liver diseases, alcohol use is linked to multiple and chronic diseases, including increased risk of cancers [4]; cardiovascular disease [5]; pancreatitis [6]; disruption in the circadian clock [7]; and impaired immune function increasing the susceptibility to bacterial and viral infections [8]. The susceptibility of patients with alcohol use disorder to develop alcohol-associated liver disease is variable indicating that, although alcohol is necessary, it is not enough to cause progressive organ dysfunction [9,10]. Consequently, factors other than the toxicity of alcohol are involved in generating health complications, one of which may be alcohol-induced changes in intestinal microbiota composition and/or function [11]. Other risk factors for progressive alcohol-associated liver disease is the amount of consumed alcohol (>1 drink/day for women, >2 drinks per day for men), drinking pattern (drinking without meal, binge drinking), genetic factors, female gender, smoking, increased body mass index and concomitant chronic liver diseases [12,13].
This review summarizes the mechanisms by which chronic alcohol intake changes the intestinal microbiota and contributes to alcohol-associated liver disease.
INTESTINAL DYSBIOSIS
The intestinal microbiota is the community of microorganisms (bacteria, archaea, fungi and viruses) that reside in the gut [14]. The human gut microbiota houses more than 10 different bacterial phyla, and there is a balance between commensal and pathogenic microbes under homeostatic conditions [15]. Dysbiosis occurs when disease or environmental factors disrupt this microbial balance contributing to the manifestation or continuation of a given disease that cannot be attributed to a single bacterial species [16,17]. Alcohol use is associated with enteric dysbiosis and intestinal bacterial overgrowth in both preclinical models and patients with alcohol abuse [18-20].
Alcohol-associated changes in the enteric microbiota are required for the development of the liver disease because intestinal decontamination with non-absorbable antibiotics (polymyxin B and neomycin) prevents alcohol-associated intestinal bacterial overgrowth and dysbiosis in mice. Importantly, reducing the intestinal bacterial burden suppressed subclinical intestinal inflammation after chronic alcohol feeding, stabilized the gut barrier and reduced ethanol-induced steatohepatitis in mice [21,22]. In ethanolfeed rats, the same antibiotic cocktail prevented liver injury and reduced the hepatic pathology score (including steatosis, inflammation, and necrosis) [23].
Interestingly, Lactobacillus was strongly suppressed and almost absent in mice fed intragastric ethanol for 3 weeks as compared with control (isocaloric) fed animals [19]. However, treatment with prebiotic fructooligosaccharides – a stimulator of beneficial bacteria growth such as lactobacilli and bifidobacteria – improved ethanol-induced steatohepatitis by inducing gene and protein expression of the bactericidal c-type lectins regenerating islet derived 3 gamma (Reg3γ) and by reducing intestinal bacterial overgrowth [19].
In humans, chronic alcohol consumption alters the composition of mucosa-associated microbiota with a lower abundance of Bacteroidetes and a higher abundance of Proteobacteria in a subset of alcoholic patients with and without liver disease compared with healthy controls [20]. While phylum Bacteroidetes is involved in carbohydrates fermentation leading to short-chain fatty acid production (mainly acetate and propionate) [24,25], Proteobacteria are known to produce lipopolysaccharides (LPS), a potent activator of the toll-like receptor (TLR)-4 [26]. The fecal microbiota of patients with alcohol use disorder and alcohol-associated liver disease was characterized by quantitative and qualitative alterations, with a reduction of bacterial diversity, reduction of Akkermansia and increase of Bacteroides. Akkermansia muciniphila produces shortchain fatty acids such as acetic acid from mucin and supplies energy to goblet cells, improving the intestinal barrier function [27]. Moreover, several reports indicate its effects on glucose and lipid metabolism, and that certain food ingredients such as polyphenols may increase its abundance in the gut [28]. In alcohol-dependent patients with high intestinal permeability, the level of Faecalibacterium prausnitzii, a bacterial species known for its antiinflammatory properties, was decreased. Conversely, those patients had higher plasma interleukin (IL)-8 levels, an inflammatory cytokine [29]. Intestinal dysbiosis has been associated with the severity of alcohol dependence and cirrhosis, and deteriorating dysbiosis is associated with cirrhosis progression [30]. Severe alcoholic hepatitis was associated with higher fecal proportions of Bifidobacteria, Streptococcus, Enterobacteria [30,31], and Enterococcus [30], and fewer proportions of Atopobium [30]. For example, differences in fecal microbiota composition were observed in patients with alcohol use disorder and alcoholic hepatitis as compared with non-alcoholic subjects [31]. In patients with alcoholic hepatitis, 5.59% of fecal bacteria were Enterococcus spp., compared with almost none in controls (0.023%) [31]. Fecal samples from patients with alcoholic hepatitis had about 2,700-fold more Enterococcus faecalis (E. faecalis) than non-alcoholic controls [31]. The exotoxin cytolysin, secreted by E. faecalis, was discovered to exert a deleterious effect on ethanol-induced liver disease in mice [31]. The presence of cytolysin-positive (cytolytic) E. faecalis correlates with liver disease severity and mortality in patients with alcoholic hepatitis [31], but no correlation was found in patients with non-alcoholic fatty liver disease [32].
Bacterial infections are a serious complication of cirrhosis, as they can lead to decompensation, multiple organ failure, and/or death. Because bacterial translocation from the gut lumen to extraintestinal sites causes infections, prevention of infections is mostly based on the use of orally administered, poorly absorbed antibiotics (known as selective intestinal decontamination) [33,34]. Several antibiotics were tested and/or used for this purpose, such as polymyxin, neomycin, gentamycin, colistin, paromomycin, and trimethoprim/sulfamethoxazole [35,36]. Currently, norfloxacin and rifaximin are the forms of selective intestinal decontamination for which there is the most evidence in cirrhosis [37-39]. However, the major drawback of routine antibiotic prophylaxis is the emergence of multidrug-resistant organisms [34]. Thus, novel therapies have been proposed based on their ability to modify the altered intestinal microbiota such as probiotics and prebiotics [19,40-45], fecal microbiota transplantation [46,47], phage therapy [31], among others. Evidence for their use in clinical practice is limited and all require further studies.
CHANGES IN MICROBIAL METABOLITES
Changes in the gut microbiota affect the host immune system by altering tryptophan metabolism [48]. Furthermore, endogenous tryptophan metabolites synthesized by the host (kynurenines, serotonin, and melatonin), and bacterial metabolites (indole, indole derivatives, skatole, and tryptamine) play an important role in regulating intestinal and systemic immune homeostasis [49]. Detailed pathways of tryptophan metabolism were recently reviewed by Hendrikx and Schnabl [50].
Several bacterial species convert tryptophan into indole and indole derivatives mainly via the enzyme tryptophanase, which is expressed in many gram-negative, as well as gram-positive bacterial species including Escherichia coli, Clostridium spp. and Bacteroides spp. [51]. Diverse indole derivatives, such as indole-3-aldehyde, indole-3-acetic acid, indole-3-propionic acid, indole-3-acetaldehyde, and indole acrylic acid bind and activate the aryl hydrocarbon receptor (AhR) [48-50]. AhR is a cytosolic ligand-activated transcription factor that is important in xenobiotic metabolism and serves as a regulator of immunity and inflammation, which involves modulating adaptive immunity and gut barrier function [48,49]. Activated AhR acts as anti-inflammatory signaling pathway that regulates the development of intraepithelial lymphocytes and innate lymphoid cells, which are important in the defense against invading pathogens and maintenance of intestinal homeostasis [52]. Moreover, AhR has been implicated in antimicrobial defense via induction of IL-22 expression by group 3 innate lymphoid cells (ILC3) [53,54]. IL-22 further regulates the microbial composition and enhances antimicrobial defense via the induction of antimicrobial proteins [52].
Dietary tryptophan supplementation altered intestinal microbial composition and diversity, improved intestinal mucosal barrier function, activated AhR signaling, and downregulated expression of inflammatory cytokines in the large intestine of weaned piglets [55]. Moreover, the metabolite indole-3-acetic acid, produced by Bacteroides spp. and Clostridium spp. [51], modulated inflammatory responses of hepatocytes and macrophages attenuating release of pro-inflammatory cytokines and cytokine-induced lipogenesis [56]. In a mouse model of ethanol-induced liver disease, ethanol-associated dysbiosis reduced intestinal levels of indole-3-acetic acid and activation of the AhR, which resulted in a decreased expression of IL-22 in the intestine and reduced expression of Reg3γ. Oral supplementation of indole-3-acetic acid protected mice from ethanol-induced steatohepatitis by inducing intestinal expression of IL-22 and Reg3γ, which prevented bacterial translocation to the liver [22]. The prevention of liver damage by non-absorbable antibiotics was associated with restored expression of IL-22 mRNA in lamina propria cells and IL-23-driven production of IL-22 by ILC3 [22], most likely by increasing intestinal levels of indole-3-lactic acid [57] and/or possibly other indole derivatives. Indole-3-lactic acid, produced by Bifidobacterium [58] and Lactobacillus spp. [57,59], have been reportedly involved in inducing immunoregulatory T cells [57] and in suppressing inflammatory T cells [60]. Indole was also shown to prevent LPS-mediated detrimental effects in the liver by downregulation of inflammatory mediators [61]. Metabolomic analysis revealed that fecal and serum levels of tryptophan were decreased in alcoholic hepatitis patients when compared with controls [62]. In line with the decrease of fecal levels of tryptophan-derived metabolites, indole-3-acetic acid, indole-3-propionic acid, and indole-3-lactic acid were also significantly reduced in alcoholic hepatitis patients [22,62].
The metabolic syndrome is associated with reduced capacity of the microbiota to metabolize tryptophan into derivatives that can activate AhR. Fecal samples of individuals with metabolic syndrome contain low levels of tryptophan-based metabolites and have reduced AhR activity. AhR ligand deficiency was also observed in mice fed a high-fat diet [63]. Treatment with either an AhR agonist or a Lactobacillus strain, to compensate for the impaired microbiota-derived AhR ligand signaling, reduces glucose intolerance and liver steatosis in animal models [63]. Depletion of indole-3-acetic acid in the liver and cecum was observed in mice fed a high-fat diet compared with those fed a regular diet [56]. This study also demonstrated that indole-3-acetic acid attenuates the release of inflammatory cytokines that induce the liver synthesis of free fatty acids, which in turn stimulate macrophages. Moreover, indole-3-acetic acid alleviates lipogenesis mediated by cytokine and free fatty acids via its direct action on hepatocytes in an AhR-dependent manner [56]. The treatment with indole-3-acetic acid attenuates high-fat diet-induced NAFLD in mice, as evidenced by amelioration of insulin resistance, lipid metabolism, oxidative stress, and inflammation [64].
In summary, fatty liver disease-associated dysbiosis results in changes of bacterial-derived metabolites, such as tryptophan metabolites. Indole derivatives are ligands for AhR and are important in immunoregulation and host defense.
IL-22
As aforementioned, gut microbiota can stimulate IL-22 expression via the production of tryptophan metabolites. IL-22, a member of the IL-10 family of cytokines, is produced by ILC3 upon stimulation and other immune cells such as Th17, Th22, natural killer cells, and γδ T cells [65]. Specific myeloid cells may also produce IL-22 under certain circumstances such as mouse macrophage-derived IL-22 produced in response to ethanol-induced cell death [66].
IL-22 acts via a transmembrane receptor complex that consists of two different subunits, IL-22R1 and IL-10R2 [67]. In hepatocytes, the biological effect of IL-22 is mediated by activation of the signal transducer and activator of transcription 3 (STAT3) and subsequent induction of anti-apoptotic and proliferative genes [68]. IL-22 is unusual among most interleukins because it does not directly regulate the function of immune cells. Rather, IL-22 targets cells at outer-body barriers, such as the skin and tissues of the digestive and respiratory systems, as well as cells of the pancreas, liver, kidney, and joints [67], controlling bacterial infection, homeostasis, and tissue repair. Thus, IL-22 is produced by immune cells and targets epithelial cells in several organs, including the intestine and the liver.
Numerous in vitro and in vivo studies have shown that IL-22 has many benefits to the liver, such as preventing hepatitis [69], stimulating liver regeneration [70], improving fatty liver [71,72], and alcoholic liver disease [68,73], and alleviating liver fibrosis [74]. In a mouse model of ethanol-induced liver disease, intestinal IL-22 has the beneficial effect of reducing bacterial translocation in the intestine by increasing the expression of Reg3γ. Bacteria engineered to produce IL-22 in the intestine (without increasing systemic IL-22) ameliorate experimental ethanol-induced steatohepatitis via induction of Reg3γ [22]. Besides, IL-22 treatment may effectively inhibit bacterial infection [75] and ameliorate kidney injury [76], two deleterious conditions that are often associated with severe alcoholic hepatitis and contribute to death of patients. More importantly, IL-22 is a promising drug for the treatment of alcoholic hepatitis because of its hepatoprotective and antifibrotic effects and relatively few know side effects during short-term use [76-78].
In addition to the direct effects on liver cells, IL-22 is a key cytokine that links intestinal immune activation to epithelial cell repair and barrier protection following damage [79]. Intestinal epithelial cells express the IL-22 receptor complex that binds IL-22 resulting in the induction of antimicrobial peptides and regenerative pathways that collectively aid in limiting bacterial invasion while promoting epithelial proliferation, wound healing, and repair [65,80,81].
ANTIMICROBIAL PEPTIDES
Chemical barriers consist of antimicrobial peptides, including defensins, lysozymes, secretory phospholipase A2, and C-type lectins, mainly involved in the segregation of gut bacteria and intestinal epithelial cells in the intestine [82]. Among those antimicrobial peptides, C-type lectins regenerating islet-derived 3 beta (Reg3β) and Reg3γ are abundantly expressed in intestinal epithelial cells and Paneth cells of the small intestine and act to maintain the inner mucus layer devoid of bacterial colonization [50,82,83]. They are upregulated upon bacterial colonization of the gut and during intestinal infection and inflammation, thereby contributing to the spatial segregation of intestinal bacteria and the epithelium [84,85]. Reg3γ is bactericidal against gram-positive bacteria by binding to peptidoglycan on the bacterial cell surface while Reg3β binds directly to LPS protecting mice against gram-negative bacteria [86-89]. Reg3β and Reg3γ also influence crypt regeneration, epithelial cell proliferation, and protect intestinal stem cells and Paneth cells from undergoing apoptosis during tissue damage [87,90,91].
Chronic alcohol consumption was associated with lower intestinal levels of Reg3β and Reg3γ in the small intestine [19,91]. Treatment with prebiotics partially restored Reg3γ protein levels, reduced bacterial overgrowth and ameliorated ethanol-induced steatohepatitis [19]. Intestinal deficiency in Reg3β and Reg3γ increases numbers of mucosa-associated bacteria, enhances bacterial translocation and promotes the progression of ethanol-induced fatty liver disease toward steatohepatitis, whereas intestine-specific overexpression of Reg3γ protects mice against it [88]. Duodenal biopsies from alcohol-dependent patients showed lower levels of Reg3γ [19] and an increased number of bacteria covering small intestinal mucosa surfaces than duodenal biopsies from non-alcoholic individuals [88]. Further, deficiency of mucin-2, the most abundantly secreted mucin in the small and large intestine, is associated with higher Reg3β and Reg3γ expression by Paneth cells and enterocytes [84], protecting mice against ethanol-induced liver disease [92]. These data indicate that antimicrobial defense plays an important role in preventing bacterial translocation and protects against alcohol-associated liver disease development [50,93,94].
BACTERIAL TRANSLOCATION
Ethanol is associated with increased intestinal permeability, defined as the passage of microbial products (including endotoxin, peptidoglycan, bacterial DNA, lipopeptides, and β-glucan) from the intestinal lumen to the mesenteric lymph nodes, and extraintestinal sites [14,95]. This increased paracellular transport across disrupted tight junctions between the enterocytes commonly occurs in patients with advanced alcohol-associated liver cirrhosis [10,96] or alcoholic hepatitis [97]. The degree of liver injury correlates with endotoxemia in patients with cirrhosis and is higher in alcoholic cirrhosis compared with other etiologies [98]. Furthermore, in patients with liver cirrhosis, endotoxemia has been associated with hepatic failure, encephalopathy, and death [99].
Results in patients with early (pre-cirrhotic) stages of alcohol-associated liver disease are mixed. Elevations in plasma LPS have been observed during the early stages of alcohol-associated liver disease [100]. However, not all heavily drinking subjects (~50%) presented with increased intestinal permeability. Indeed, patients with dysbiosis had higher intestinal permeability while patients without microbial alterations did not, despite heavy chronic alcohol consumption. Three weeks of alcohol cessation reversed changes in intestinal permeability [29,94]. This raises the possibility that only a subset of heavy drinking patients has increased intestinal permeability.
Increased intestinal permeability is a common feature in preclinical models of ethanol-induced liver disease [101]. In addition, microbial derived products appear to play an important role for ethanol-induced liver disease in mice. Mice with genetic deletions in the LPS signaling pathway are resistant to alcohol-induced liver damage [102]. Despite endotoxemia occurring in preclinical mouse models of ethanol-induced liver disease, LPS alone is not enough to result in liver injury and steatosis. We have demonstrated in three independent mouse studies that blocking translocation of viable bacteria to the liver is sufficient to reduce ethanol-induced liver disease despite the same systemic LPS level [31,88,103].
Ethanol impairs the expression of intestinal antimicrobial proteins, which induces a quantitative increase of bacteria in the mucus and epithelial cell layer. This facilitates translocation of viable bacteria from the gut lumen to mesenteric lymph nodes and the liver by mechanisms that are poorly understood and deserve future investigation. The most common bacteria involved in translocation of viable bacteria are derived from the family of Enterobacteriaceae (Escherichia coli, Klebsiella, etc.), Enterococcus and Streptococcus, while anaerobic microorganisms are rarely responsible [104]. In mice, chronic ethanol administration changes bacterial alpha diversity in the ileum, largely driven by an increase in gramnegative bacteria. Moreover, gram-negative Prevotella not only increased in the mucus layer of the ileum but also in liver samples suggesting that translocation of viable bacteria to the liver might be associated with microbiota changes in the distal gastrointestinal tract [101]. Gastric acid suppression by proton pump inhibitor increases intestinal Enterococcus and its translocation to the liver, exacerbating ethanol-induced liver disease both in mice and humans [103]. E. faecalis was detectable in the liver of mice given cytolytic and non-cytolytic E. faecalis and fed an ethanol diet, but not when mice were fed a control diet indicating that ethanol-induced changes in the gut barrier are necessary for the translocation of cytolytic E. faecalis. Transplantation of feces from cytolysin-positive patients increased translocation of cytolytic E. faecalis to the liver after ethanol administration. Bacteriophages that target cytolytic E. faecalis reduced translocation of cytolysin to the liver and abolished ethanol-induced liver disease in humanized mice [31]. These examples illustrate that translocation of viable bacteria from the gut lumen is sufficient and necessary for ethanol-induced liver disease in mice.
ALCOHOL-ASSOCIATED LIVER DISEASE
When microbial products or viable bacteria reach the liver, activation of the TLRs and the cytosolic nucleotide-binding oligomerization domain-like receptors present in both parenchymal and non-parenchymal cells occur [10,77]. LPS stimulates the innate immune response through the binding to TLR4 and its co-receptor cluster of differentiation 14 (CD14), activating, in turn, NF-κB and IL-6/STAT3 signaling in Kupffer cells, macrophages, and hepatic stellate cells [105]. TLR4 signaling is required for liver steatosis, inflammation, and a fibrotic response after chronic alcohol intake [106]. Mice with mutant TLR4, deficiency of its cellular co-receptor CD14 [107] or with inactivation of Kupffer cells are protected from ethanol-induced liver injury [102,108]. Additionally, alcohol induces LPS binding protein, TLR4 and CD14 expression thus enhancing responsiveness to endotoxin [108,109]. TLR4 activation in Kupffer cells stimulates the production of cytokines (IL-1β and tumor necrosis factor [TNF]-α), chemokines, reactive oxygen species, and leukotrienes which leads to T lymphocyte and neutrophil recruitment, hepatic stellate activation, and collagen production [106,110]. Additionally, patients with alcohol dependence have elevated plasma peptidoglycans level and increased mRNA expression of IL-1β, IL-8, and IL-18 in peripheral blood mononuclear cells. Induction of these cytokines was likely related to increased expression and activation of TLR2 receptors as well as activation of the transcription factor AP-1 and the NLRP3 inflammasome, contributing to inflammation in the liver and progression of the disease [29].
Hepatic non-immune cells, such as endothelial cells and hepatic stellate cells, also respond to bacterial products through TLRs releasing inflammatory cytokines and chemokines including IL-1, IL-6, and TNF-α as well as profibrogenic cytokines including transforming growth factor (TGF)-β1 [110]. TGF-β1, a key activator of hepatic stellate cells, can upregulate the synthesis of some extracellular matrix proteins and the cellular receptors of several matrix proteins to further promote hepatocyte injury and death in the liver [111]. Ethanol-induced oxidative stress in the liver and ethanol metabolites (acetaldehyde and adducts such as malondialdehyde or 4-hydroxynonenal) also sensitize hepatic stellate cells to activation by LPS, which results in liver injury and fibrosis after the combination of chronic alcohol feeding and LPS [112].
We have recently demonstrated that cytolysin secreted by E. faecalis can directly kill hepatocyte and promote ethanol-induced liver disease in mice [31]. All these processes together can ultimately culminate in hepatic injury and systemic inflammation contributing further to immune disarray, predisposing individuals to complications such as infections and hepatic decompensation (Fig. 1) [113,114].
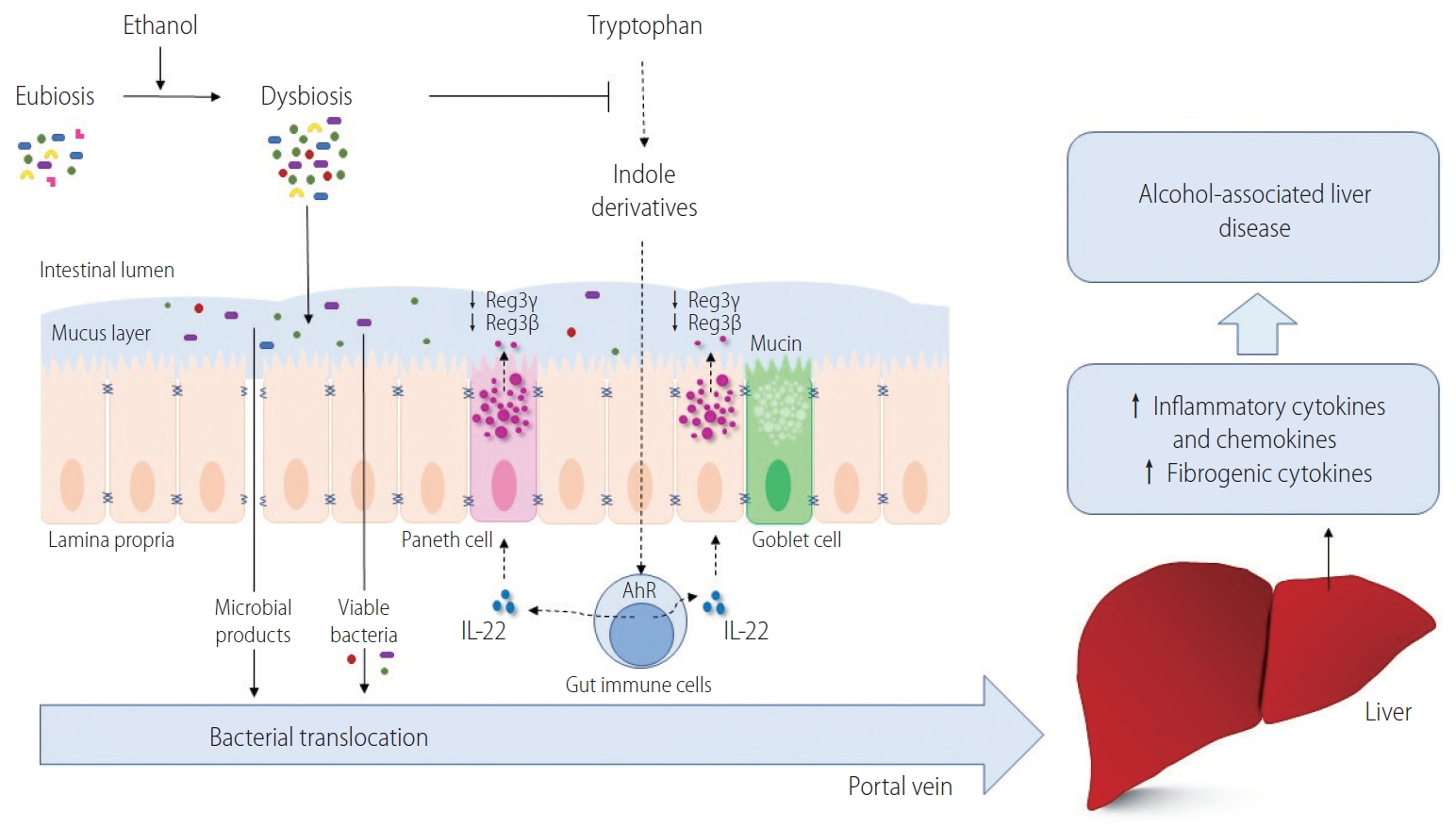
The mechanism by which chronic alcohol intake changes the intestinal microbiota and contributes to alcohol-associated liver disease. Alcohol abuse is accompanied by intestinal dysbiosis and bacterial overgrowth. Certain gut bacteria can metabolize tryptophan into indole derivatives that can bind and activate the AhR. Activated AhR induces the expression of IL-22 in lamina propria immune cells, which stimulates mucosal defense via the production of antimicrobial peptides, such as Reg3β and Reg3γ, in intestinal epithelial cells and Paneth cells. Intestinal deficiency of Reg3β and Reg3γ increases the numbers of mucosa-associated bacteria and facilitates bacterial translocation through the portal vein. In the liver, viable bacteria and microbial products induce hepatic inflammation, hepatocyte death and fibrotic responses. Reg3γ, regenerating islet-derived 3 gamma; Reg3β, regenerating islet-derived 3 beta; IL-22, interleukin-22; AhR, aryl hydrocarbon receptor.
CONCLUSIONS
We have considerably increased our knowledge about how chronic alcohol intake changes the intestinal microbiota and contributes to disease progression. Gut dysbiosis and bacterial translocation result in immune activation and hepatic injury. Therefore, restoration of intestinal eubiosis can be used as prevention and therapeutic approach. This could be achieved by manipulating of gut microbiota through dietary changes, probiotic, prebiotic or symbiotic therapy. A promising therapeutic approach is the modification of the tryptophan-aryl hydrocarbon receptor pathway to increase the intestinal expression of IL-22 and antimicrobial proteins like Reg3γ and Reg3β. Genetically engineered bacteria producing specific factors to restore this pathway, might have fewer side effects than systemic administration. A small clinical trial showed promising results using an IL-22 agonist in patients with alcoholic hepatitis. Finally, editing the microbiota with bacteriophages is another therapeutic modality that could be explored to reduce the progression of alcohol-associated liver disease. Future clinical trials are required to test these possibilities.
Notes
Authors’ contribution
BGM drafted the manuscript; BS edited and revised the manuscript; both approved the manuscript’s final version.
Conflicts of Interest: B.S. has been consulting for Ferring Research Institute, Intercept Pharmaceuticals, HOST Therabiomics and Patara Pharmaceuticals. B.S.’s institution UC San Diego has received grant support from BiomX, NGM Biopharmaceuticals, CymaBay Therapeutics, Synlogic Operating Company and Axial Biotherapeutics.
Acknowledgements
This study was supported in part by NIH grants R01 AA020703, R01 AA24726, U01 AA026939, and services provided by P30 DK120515 and P50 AA011999.
Abbreviations
AhR
aryl hydrocarbon receptor
CD14
cluster of differentiation 14
E. faecalis
Enterococcus faecalis
IL
interleukin
ILC3
group 3 innate lymphoid cells
LPS
lipopolysaccharides
Reg3β
regenerating islet-derived 3 beta
Reg3γ
regenerating islet-derived 3 gamma
STAT3
signal transducer and activator of transcription 3
TGF
transforming growth factor
TLRs
toll-like receptors
TNF
tumor necrosis factor