


INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is a complex disorder that affects a large proportion of the world population of all ages [1]. The disease pathogenesis involves a myriad of factors, including genetic susceptibility and predisposing metabolic comorbidities, such as obesity and type 2 diabetes, as well as environmental exposure and lifestyle, which jointly shape the NAFLD epigenome [2-4].
Yet, despite more than two decades of extensive research in the field of NAFLD, there is currently no approved therapy for nonalcoholic steatohepatitis (NASH)ŌĆöthe severe histological form of the disease. Moreover, none of the new drugs or small molecules with diverse molecular targets on the pathways of hepatocyte injury, inflammation, and fibrosis can achieve primary efficacy endpoints [5-10]. Diverse factors have been postulated to contribute to the low success rate in NAFLD/NASH drug discovery, including lack of robust animal models needed for preclinical studies, insufficient target engagement or target modulation by the novel drugs, absence or insufficient demonstration of a proof-of-concept in early trials, and/or high false discovery rate (FDR) in phase 2 trials [11].
Likewise, it has been hypothesized that, as data on the candidate drugs are not only insufficient but are also not corroborated by genetic inactivation, pharmacological inhibition, antisense oligonucleotides, and/or small interfering RNAs, this poses an additional obstacle to achieve consistent and sustained effects on severe histological outcomes, including improvement in fibrosis scores [11].
The aforementioned pitfalls could potentially be overcome through systems biology analysis, aiming to integrate knowledge of signaling pathways [12], the genetic information of susceptibility genes [4], and multiple tissue-specific OMICs-related experiments that include large-scale transcriptomic, proteomic, and metabolomic profiles [13,14], and more recently metagenomics of the liver tissue [15].
Furthermore, the approach founded on precision medicine is expected to enhance the effectiveness of novel therapies, including elucidation of predictors of drug response. Hence, this review summarizes significant trends and developments in precision medicine with focus on new potential therapeutic discoveries modeled by systems biology approaches.
THE PATH TOWARDS PRECISION MEDICINE: A SHORT CONCEPTUAL APPRAISAL
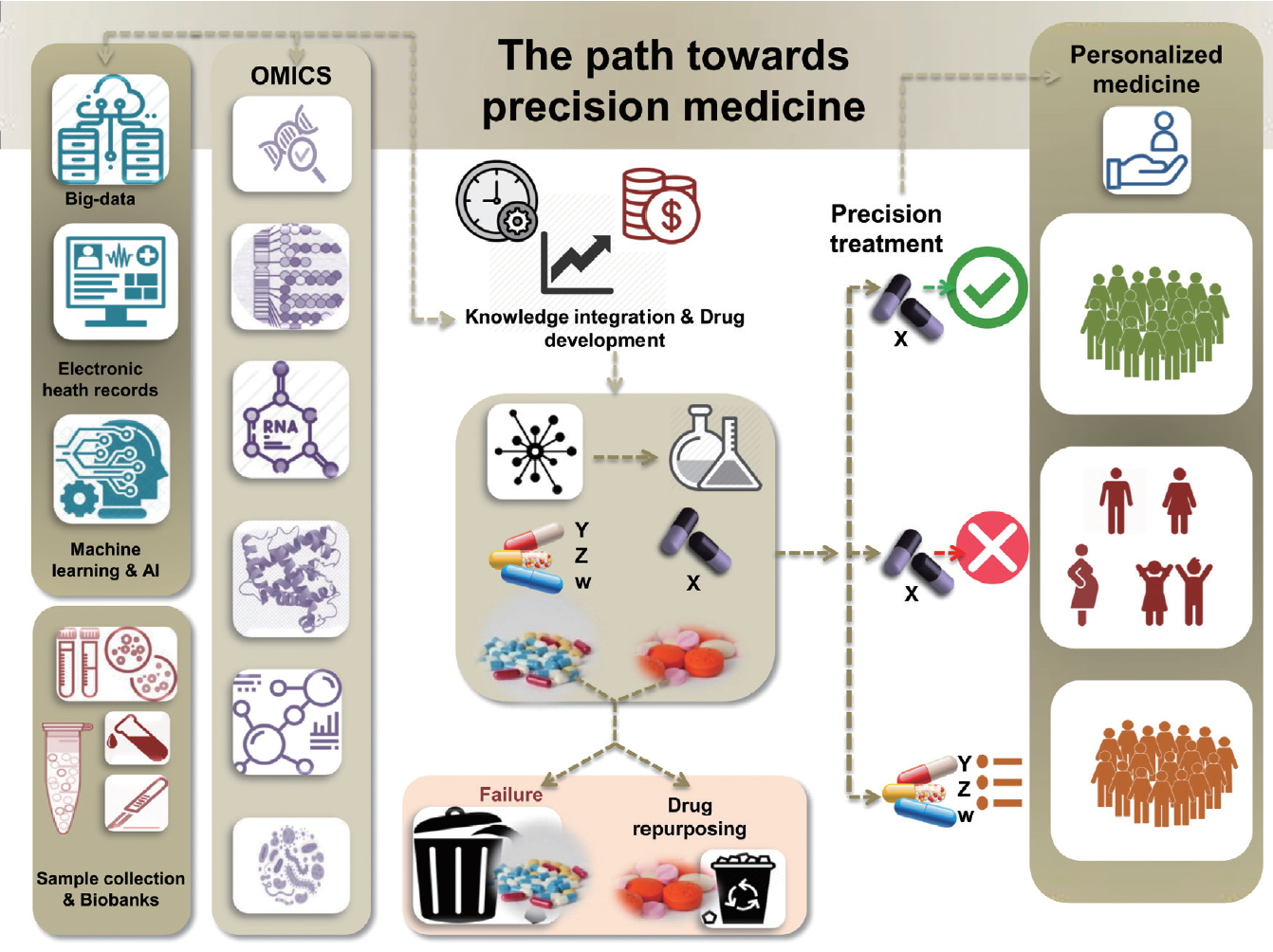
The ultimate goal of precision medicine is to develop precision treatment strategies that rely upon a holistic understanding of differences in genetic and underlying molecular pathogenic factors, as well as responses to environmental stressors, among patients. Figure 1 describes milestones in the path towards precision medicine, which include the integration of big data, comprising information from electronic health records of thousands of patients and machine learning strategies, such as artificial intelligence, to assist with the development of algorithms for combining and decoding such complex information. In addition, collections of biological samples in large biobanks linked to patient data increase the likelihood of finding robust disease pathogenesis signatures derived from OMICs state-of-the-art approaches. Knowledge integration and data modeling and analysis are vital processes at the interface with drug discovery. It should be emphasized that time and cost are presently the key limiting factors at these stages. However, as the technological advances progress further, it is expected that, in the near future, the gap between the discovery of potential drugs and clinical validation will be considerably narrowed. Still, even when complex algorithms aimed at multi-scale modeling of OMICs data succeed in identifying a potential drug target, only some of these medications will eventually be included into primary or secondary therapeutic protocols for treating the target disease. At this stage, at least three clinical scenarios will likely emerge. One possibility is that a putative drug ŌĆ£xŌĆØ demonstrates not only a good safety profile but also succeeds in achieving the objective endpoints in a large group of patients (Fig. 1). Another possibility is that, in a small group of patients, ŌĆ£xŌĆØ would be contraindicated for diverse reasons, including safety concerns in some vulnerable groups, such as children, pregnant women, or patients suffering from chronic kidney disease, advanced cardiovascular disease, etc. It is also possible that certain patients will require a different kind of drug because their underlying molecular and/or genetic profile does not align with the molecular profile of the drug ŌĆ£xŌĆØ (Fig. 1).
In addition, pathway-derived drugs may emerge from drug repurposing. A comprehensive algorithm for drug repurposing or repositioning in NASH, which primarily relies on identifying and developing new uses for existing drugs, was recently published [16]. In fact, drugs which had an acceptable safety profile, but failed in achieving the expected response for some diseases, could be used to treat a different condition. It is also noteworthy that a large number of discovered drugs fail and never pass preclinical testing. Hence, the ultimate role of precision therapy relies on defining appropriate prediction strategies for implementing patient-based therapies.
PRECISION MEDICINE AND LARGE-SCALE GENOME AND EXOME SEQUENCING DATA
Extant studies on the genetic component of NAFLD indicate that, after 12 years following the discovery that an allele in a patatin-like phospholipase domain containing 3 (PNPLA3) variant (rs738409 [G], encoding 148M) was associated with increased hepatic fat [17] and NAFLD disease severity [18], knowledge of the disease heritability is still incomplete [3,4,12]. The correlation between rs738409 and the risk of developing fatty liver, NASH, and fibrosis is perhaps one of the strongest worldwide-replicated effects for a common variant modifying the individual susceptibility of NAFLD and NASH (explaining ~5.3% of the total variance) [3,4,18,19]. Indeed, available evidence indicates that homozygous carriers with the G-risk allele of rs738409 present 3.24-fold greater risk of higher liver necroinflammatory scores and 3.2-fold greater risk of developing fibrosis when compared with homozygous CC carriers [19].
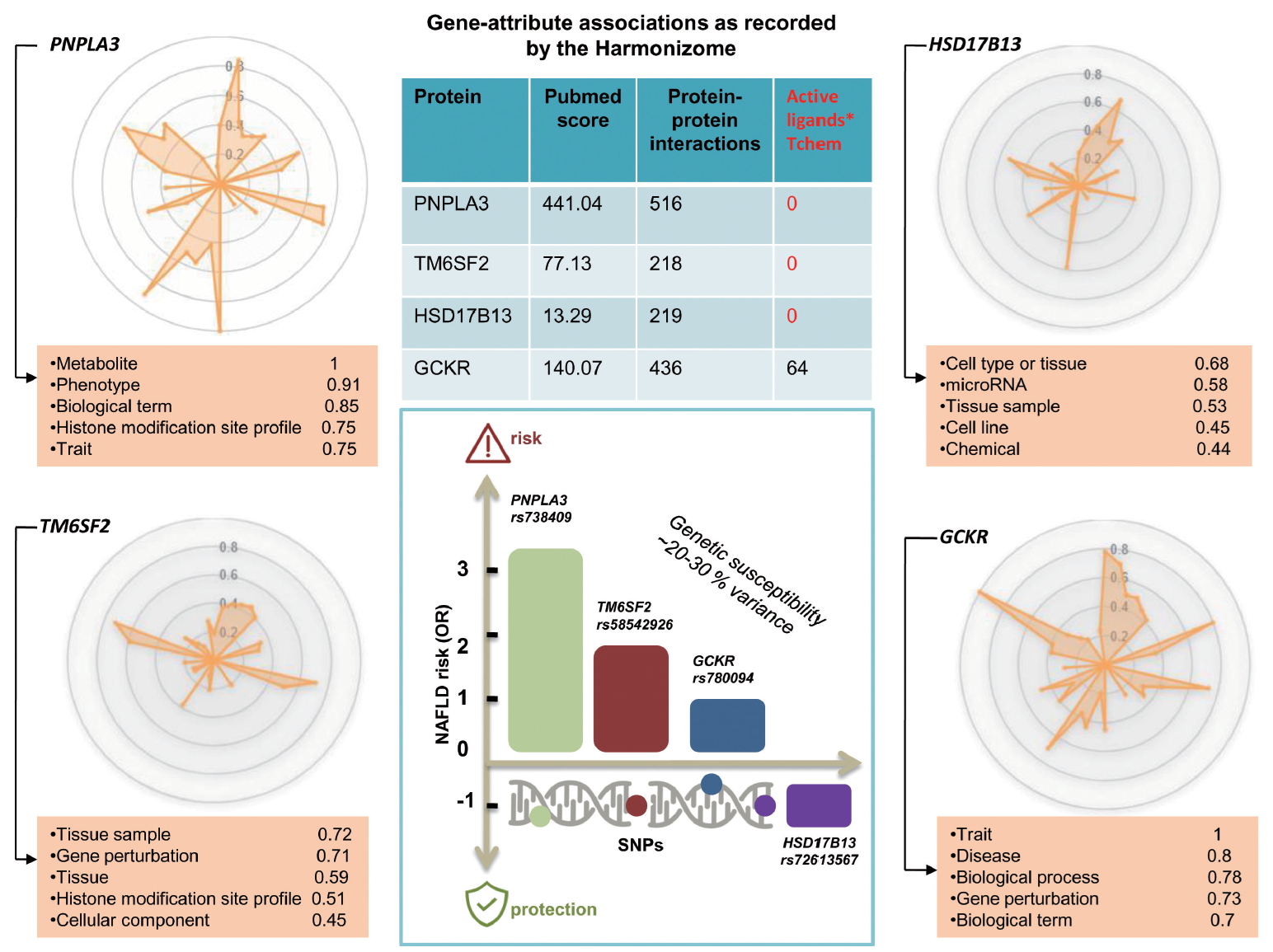
Findings yielded by genome-wide association studies (GWAS) as a part of which the heritability of hepatic steatosis was explored at the population level and/or NASH was examined in patients with liver biopsy, consistently show that at least four loci (PNPLA3, transmembrane 6 superfamily member 2 [TM6SF2], glucokinase regulator [GCKR], and hydroxysteroid 17-beta dehydrogenase 13 [HSD17B13]) [17,20-22] are involved in the genetic susceptibility of the disease (Fig. 2). Conflicting results have been published regarding the rs641738 C/T located in transmembrane channel-like 4 (TMC4) exon 1 (p.Gly17Glu) and 500 bases downstream of the membrane bound O-acyltransferase domain containing 7 (MBOAT7; TMC4/MBOAT7), which were initially described in Italian population [23] but could not be replicated in other populations around the world [24-26], including a large cohort of European patients partaking in a GWAS, for whom NAFLD was diagnosed by liver biopsy [27].
Targeting disease-associated genes has been proven successful in the treatment of numerous human diseases, cancers in particular. In this field, target drugs are associated with the effect of a mutant protein and/or are designed to interfere at the gene or protein level. However, for this goal to be fully realized, full knowledge of the target genes/proteins is required, not only at all molecular levels, but also at the level of chemical-gene/protein interactions, which would ultimately allow identification of potential active ligands.
Likewise, drug discovery and precision medicine require complete understanding of new technologies as well as gene and protein biology of the selected target. Radar plots depicted in Figure 2 show the discrepancy in the level of knowledge on the four loci reproducibly implicated in the biology of NAFLD and the disease severity. For example, the plots show that, while extensive knowledge on PNPLA3 at different levels already exists, ranging from metabolic aspects to epigenetic information and traits associations, that pertaining to HSD17B13ŌĆöthe newly discovered gene with a putative loss-of-function variant implicated in protective effects against NASH and severe histological stages [20,28]ŌĆöis limited at all levels of gene and protein biology (Fig. 2). The rs72613567 insertion/deletion variant, the functional consequence of which is a splice donor variant of the HSD17B13 [20], represents an interesting model of a candidate molecule for treating NASH and fibrosis [29]. In fact, although the information about druggable binding domains in HSD17B13 is scarce due to the lack of an experimental 3D structure, other members of the protein family, such as HSD17B11 with high homology, present putative binding pockets for small molecules [30].
As indicated in the PNPLA3 illumination graph, considerable knowledge has been accumulated on the gene and protein, including gene-attributed associations, protein interactions, and high PubMed score; yet, no information on the active ligands of the protein is currently available (Fig. 2).
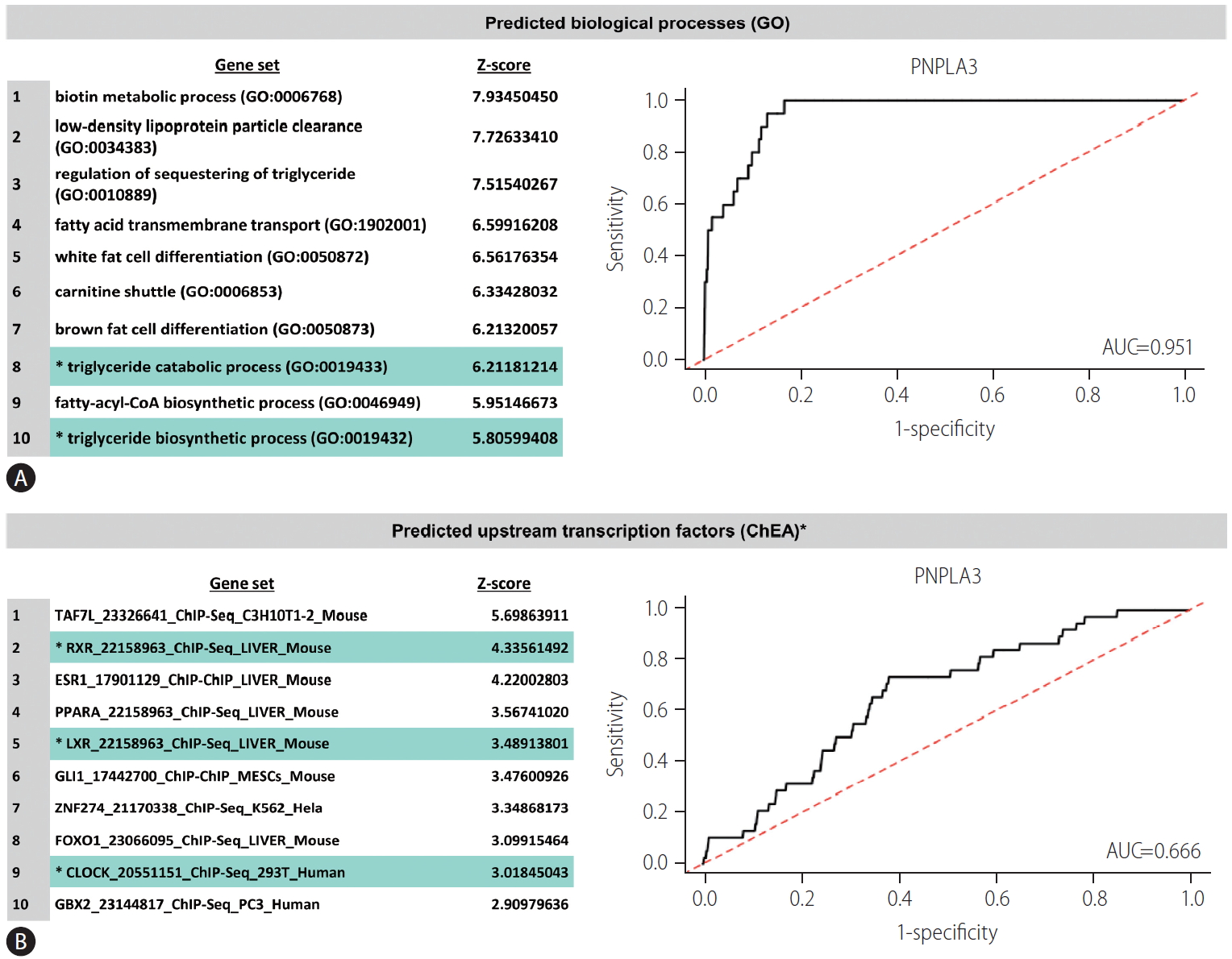
The protein encoded by PNPLA3 is a triacylglycerol lipase that mediates triacylglycerol hydrolysis, mostly in adipocytes [12]. The encoded protein, which appears to be membrane-bound, may be involved in the energy usage/storage balance in adipocytes [12]. Figure 3A shows area under the receiver operating characteristics (AUROC; 0.951) for predicted functional processes linked to PNPLA3, including triglyceride catabolic process (gene ontology [GO]: 0019433) and triglyceride biosynthetic process (GO: 0019432) (prediction was done by the Harmonizome). Importantly, extensive mining of publicly available RNA-seq data acquired through human and mouse experiments (https://amp.pharm.mssm.edu/archs4/) uncovered interesting upstream transcription factors (AUROC, 0.666) in Figure 3B, including RXR (retinoid x receptor), LXR (liver x receptor, nuclear receptor subfamily 1 group H member 3), and CLOCK (circadian locomotor output cycles protein kaput, formally known as circadian clock regulator). The protein encoded by the CLOCK plays a central role in the regulation of circadian rhythms. In our previous publications, we reported for the first time that CLOCK genetic variation is associated with obesity and NAFLD [31,32]. The haplotype of rs1554483G and rs4864548A was found to be associated with a 1.8-fold risk of overweight status or obesity [32], whereas rs1554483 was shown to be associated with all histological traits of NASH, including fibrosis [31]. Considered jointly, this body of evidence suggests that the putative cross-talk between PNPLA3 and CLOCK could explain the link among NAFLD genetic susceptibility, the environment, and the circadian regulation of liver metabolism. However, further experiments are required to prove this hypothesis.
TM6SF2, of which rs58542926 C/T (E167K) was initially associated with liver fat accumulation and aminotransferase levels in a large GWAS study [21] and further replicated in subsequent studies [21,33-35], encodes for a protein involved in lipid metabolism [12]. Diverse areas of molecular knowledge gained on this gene/protein are presented in Figure 2.
Radar plot of GCKR presented in Figure 2, of which rs780094 presents a very modest [36] effect (odds ratio [OR], 1.2) on NAFLD biology [3,12], and shows that considerable gene/protein knowledge, including at least 64 putative ligands, has been already acquired in this domain [4].
Precision medicine has emerged as a result of comprehensive knowledge of the druggable genome/proteome. Thus, advancing the chemical genetics research, which is based on the screening of low-molecular weight compounds that act by binding to specific receptors/proteins, is crucial to move this promising research domain forward in the right direction. It is worth mentioning that the incomplete knowledge on the druggable genome of NAFLD/NASH severely undermines the drug discovery progress and reduces the chances of having robust and safe drug candidates. Therefore, the substantial gap between the knowledge of NAFLD-predisposing genes and that related to putative protein ligands needs to be urgently addressed.
NAFLD AND THE PUTATIVE CLINICAL BENEFITS OF POLYGENIC RISK SCORES (PRSs)
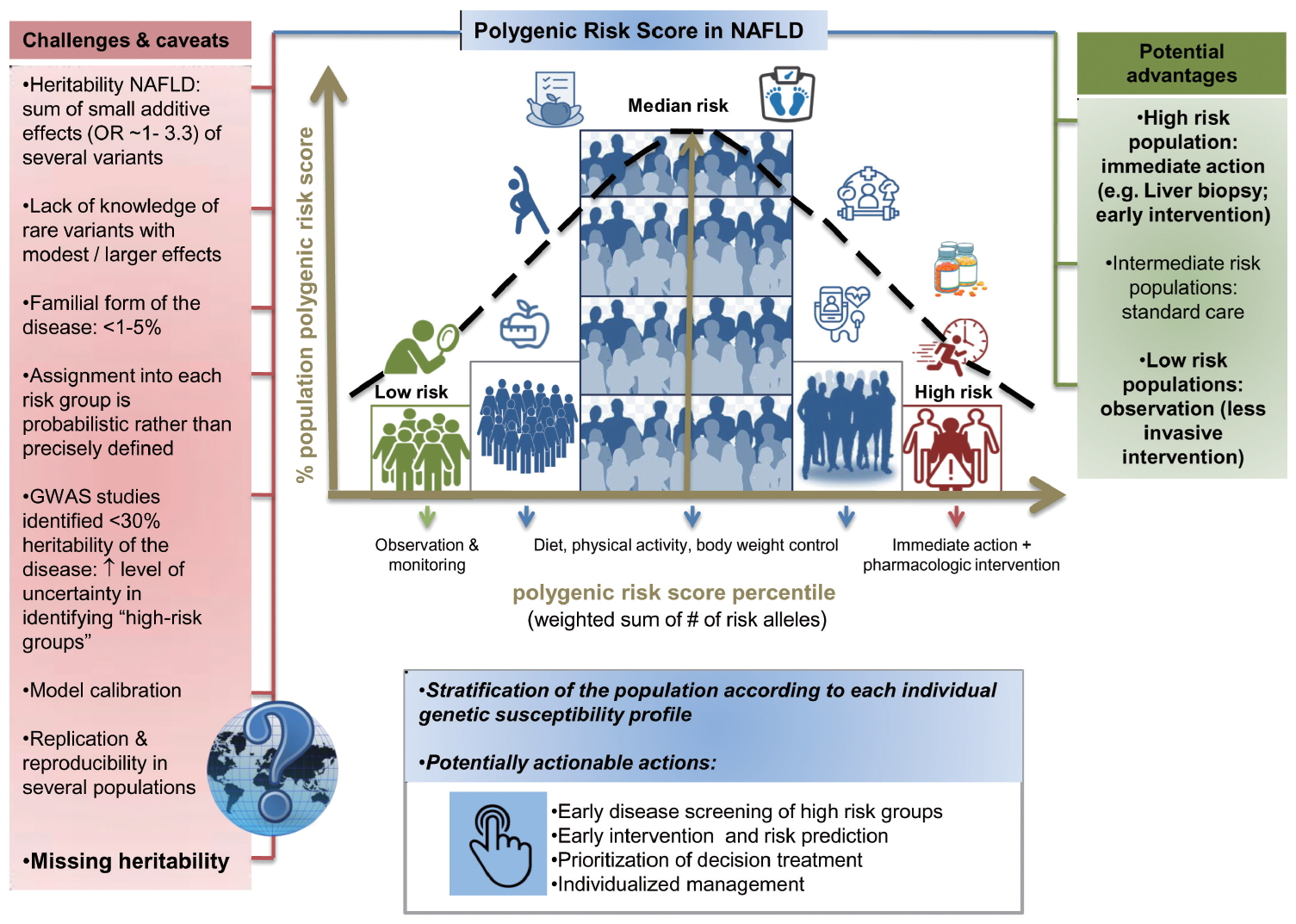
Estimating the susceptibility risk of a given patient to develop a particular disease and/or to progress into severe disease stages is the ultimate aim of precision medicine. Most researchers concur that the PRS distribution, which is based on the sum of all independent risk single nucleotide polymorphisms (SNPs; ideally weighted by their size effects in a given population), could be approximated by the Gaussian (normal) curve (Fig. 4). PRSs are theoretically designed to explain the relative risk of a disease, as these scores provide information on how a person compares with others with different genetic susceptibility background. However, PRSs do not necessarily follow normal distribution, due to several factors, including differences in the population structure or admixture.
In the case of NAFLD and NASH, PRSs could be conceptually very advantageous not only for allowing early disease detection, but also for implementing timely actionable measures (Fig. 4). For example, invasive diagnostic approaches, such as liver biopsy, as well as early pharmacological intervention, would be advised for high-risk populations (those at the right-tail of the PRS distribution curve pertaining to the relevant population) whereas low-risk individuals (i.e., those on the left-tail of the curve) would be monitored until clinical risk becomes evident (Fig. 4). For those deemed at low or medium risk, which probably applies to the large majority of the affected patients, lifestyle changes would be advised, including regular physical activity and dietary modifications aimed at optimizing body weight and controlling the key metabolic risk factors (lipid traits and glucose metabolism).
Despite these benefits, several important concerns related to the clinical implementation of PRSs also exist, as noted in Figure 4. In particular, use of PRSs in clinical settings will remain impractical until the heritability of NAFLD is fully elucidated, and rare and familial forms of the disease are revealed. Inclusion of additional genetic information will certainly aid in overcoming these issues as the predictive power of PRSs improves and the proportion of individuals at risk diminishes.
For instance, early detection of a rare nonsense GCKR mutation (rs149847328, p.Arg227Ter) in a NAFLD patient with associated comorbidities, including morbid obesity and type 2 diabetes, when combined with prompt pharmacological intervention, could potentially prevent or even reverse the disease progression into liver cirrhosis [37].
Furthermore, the currently available knowledge on the reproducibility and replication of genetic variants of NAFLD across diverse populations around the world is insufficient (Fig. 4), and the effect sizes of most of the variants are yet to be established. Indeed, in the large majority of GWAS focusing on NAFLD, the genetic susceptibility in patients of European ancestry has been examined. This creates a considerable gap in extant knowledge, as SNP-based information on Caucasian and/or people of European descent may not be relevant for inferring the relative risk of NAFLD in non-European populations. Significant efforts have been made, however, to overcome this limitation. For example, Kawaguchi and coworkers conducted a GWAS in Japanese population and demonstrated that patients with NASH are genetically and clinically different from other population subgroups [38]. In addition, as a part of a large population-based GWAS that involved 1,593 patients and 2,816 controls, Chung and coworkers characterized the genetic profile of Korean NAFLD patients [39]. With the exception of these remarkable examples from Asia, the percentage of non-European ancestry population in NAFLD GWAS studies, including those of African descent and/or other ethnic minorities, is dramatically low.
In addition to these shortcomings, certain technical issues, such as model calibration and calculation algorithms, must be overcome to fully benefit from the PRS implementation.
Genetic markers are already being used as tools for personalizing clinical practice, including treatment decisions [4]. Nevertheless, the utility of genetic variants in NAFLD risk estimation remains inferior to classical predictive or imaging approaches, as explained earlier [4], In fact, knowledge of population structure and global heterogeneity of variants implicated in the disease progression is rather limited.
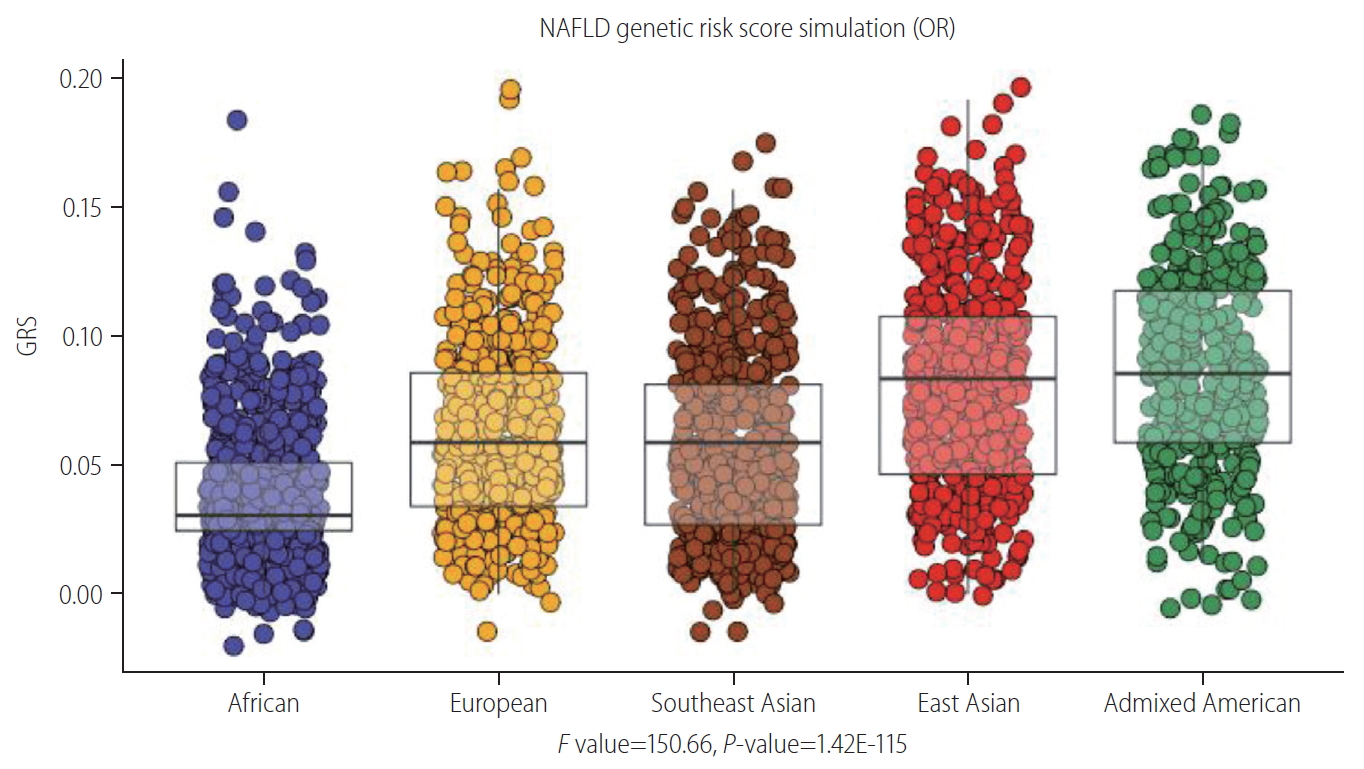
Thus, to simulate the potential utility of a NAFLD-PRS we used population-specific distribution information on the four aforementioned SNPs (PNPLA3-rs738409, TM6SF2-58542926, HSD17B13-rs72613567, and GCKR-780094) and the intergenic variant LYPLAL1-rs12137855 [22]. To compute the NAFLD-PRS, we used the GlobAl Distribution of GEnetic Traits (GADGET) web server available at https://gadget.biosci.gatech.edu/compute/; the formula used to compute the score is based on the original description of Chande et al. [40]. The GADGET web server provides access to publicly available genotype data sourced from the 1000 Genomes Project (1KGP) Phase 3 data release and individual trait SNP sets parsed directly from the NHGRI-EBI GWAS Catalog annotations (https://www.ebi.ac.uk/gwas/) [40]. Box plots representing NAFLDPRS distribution by five major continental groups (Africa, Europe, South and East Asia, and America) are shown in Figure 5. According to this information, the overall predicted risk to individuals entailed by the presence of NAFLD-implicated variants in their genomes is about ~0.20. Nevertheless, the PRS or genetic risk score (GRS) reflects disparities in NAFLD risk levels across different populations (Fig. 5), which may be due to the disparities in genetic knowledge or may indicate real differences in the genetic risk. Thus, our analysis emphasizes the potential caveats of implementing this strategy globally. Of course, our simulation-based approach, which is based on information available in public databases, did not allow us to control for all possible effects, including demographic variables and/or other population risk estimates. Nonetheless, the GRS presented here provides a good starting point for illustrating the current situation, as well as for further investigations aimed at closing the gap in the knowledge needed to generate valuable advances in this field. It is worth noting that, as is the case for almost all human traits, variance in the NAFLD genetic risk within each population is much greater than among continental groups. This fact does not, however, imply that there is no continental group-specific risk profile. Available evidence indicates that certain trends exist at the global level, whereby the lowest GRS is associated with African population, intermediate GRS relates to European and Southeast Asian, and the highest GRS to East Asian and Admixed American groups.
NAFLD GENES, PLEIOTROPIC RELATIONSHIPS, AND PRECISION MEDICINE
Available evidence suggests that genetic factors associated with NAFLD exhibit similar patterns of correlation with genetic factors related to other complex diseases [4]. For instance, findings yielded by large GWAS studies indicate that PNPLA3 -r738409 and TM6SF2-rs58542926 are associated with extra-hepatic traits, including hematological (plateletcrit and count) and lipid traits, and some other interesting pharmacogenetic associations (http://www.phenoscanner.medschl.cam.ac.uk/) (Fig. 6). Importantly, the minor allele frequency of these two variants across different populations supports the prevalent view that the major genetic modifiers of NAFLD are likely ancestry-specific. Therefore, this observation should be specifically examined when designing precision medicine strategies.
The genetic pleiotropy between the aforementioned variants and non-liver related traits includes known NAFLD-associated comorbidities, such as cardiovascular risk. Phenotypic covariation presents not only significant challenges in clinical practice but also imposes tremendous constraints on identifying novel therapeutic targets. The clinical paradox of TM6SF2ŌĆÉrs58542926 C>T is a clear example of that. The C (Glu167) allele has been consistently associated with increased cardiovascular risk [41], and the T allele (Lys167) is known to be associated with a higher risk for NAFLD and NASH [21,33,42,43]. These opposite effects are dependent on circulating and liver triglyceride levels, respectively. Consequently, TM6SF2 does not seem to be a useful drug target because any impact on the protein would eventually lower blood lipids, which will in turn reduce the risk of myocardial infarction, while simultaneously increasing the risk of developing NAFLD [42].
As explained in previous paragraphs, drug development is a long process characterized by highly uncertain outcomes. At present, 10ŌĆō15 years typically elapse between target discovery and clinical application. Hence, some computational solutions, including modeling and over-representation analysis grounded in systems biology, may assist with more accurate prediction of drug candidates based on disease-associated genes/proteins. To illustrate this concept, we employed two different strategies. First, we leveraged existing information on the molecular targets (genes/proteins) involved in NAFLD/NASH pathogenesis and performed over-representation analysis using a drug-related functional database. The training set of genes/proteins was obtained by literature-data mining offered by the Genie web server (http://cbdm01.zdv.uni-mainz.de/~jfontain/cms/?page_id=281)ŌĆöa tool that computes associations of genes with diseases using biomedical literature annotations. Using this approach, 901 abstracts from PubMed were retrieved using the search terms ŌĆ£fatty liverŌĆØ and ŌĆ£humanŌĆØ (taxonomic identifier 9606; no literature extension by orthology) with the abstracts from the all PubMed database serving as the background set. Supplementary Table 1, intended for online publication only, shows the final ranked list comprising of 938 genes/proteins.
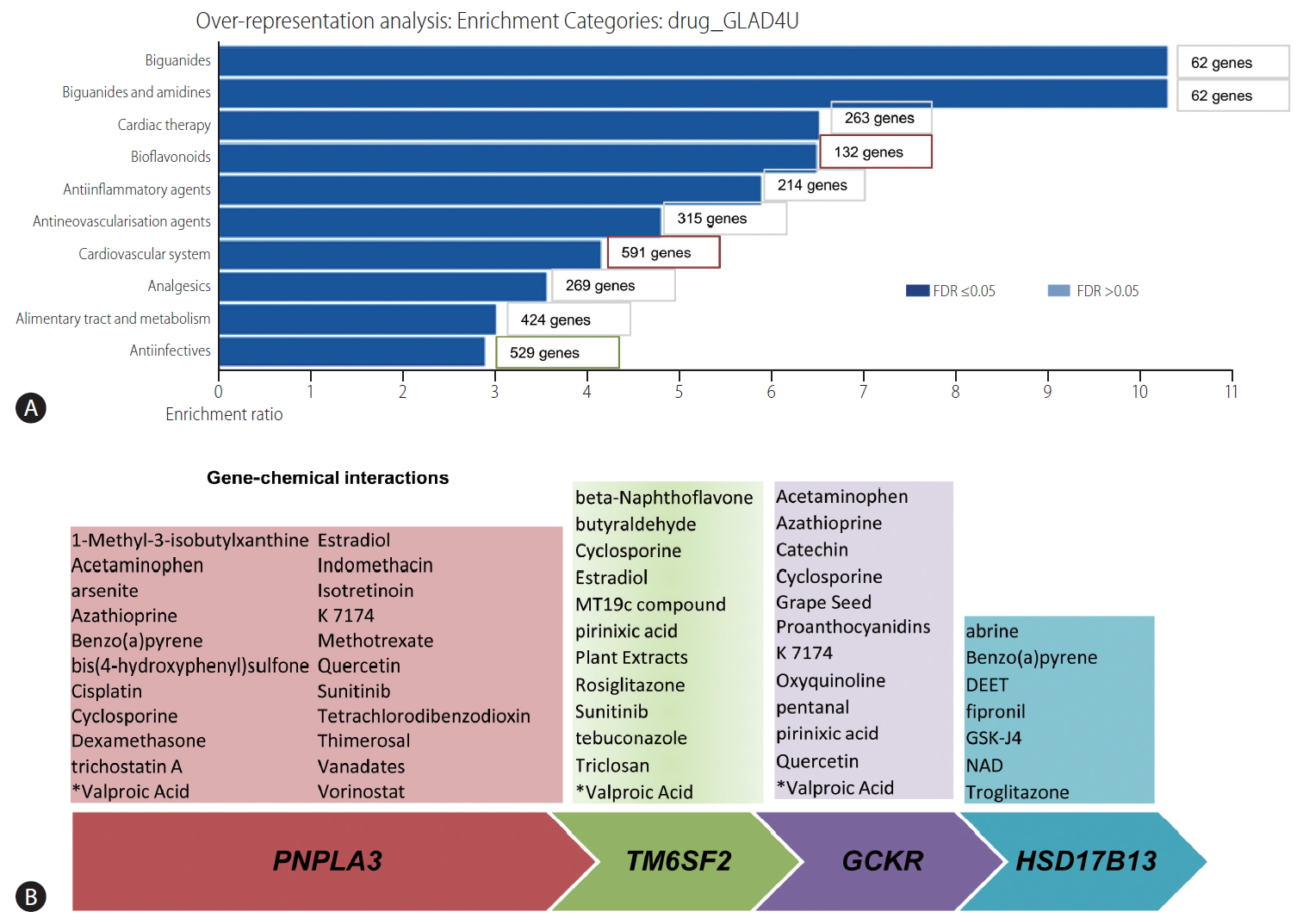
The aforementioned list of retrieved genes/proteins was used to perform over-representation analysis in the webserver WebGestalt (WEB-based Gene SeT AnaLysis Toolkit)ŌĆöa functional enrichment analysis tool available at http://www.webgestalt.org/. Drug terms were downloaded from ParmGKB by the WebGestalt, and individual drug terms associated with genes were inferred using the GLAD4U option. Accordingly, the drug enrichment results based on the NAFLD training set are shown in Figure 7A. The bar chart shows ten categories that passed the FDR <0.05, whereby cardiac therapy and cardiovascular system were overrepresented by the largest number of genes (263 and 591, respectively). Other expected drug categories are anti-inflammatory agents and biguanides, which are probably justified by the significant enrichment of genes/proteins associated with inflammation and glucose metabolism in the training set. In fact, the top ten genes/proteins of the training list were ADIPOQ (adiponectin), PNPLA3, PPARG and PPARA (peroxisome proliferator-activated receptor gamma and alpha), FGF21 (fibroblast growth factor 21), RBP4 (retinol binding protein 4), GPT (glutamic-pyruvic transaminase), SREBF1 (sterol regulatory element binding transcription factor 1), LEP (leptin), and NR1H3 (nuclear receptor subfamily 1 group H member 3, also known as liver X receptor alpha). Lastly, the analysis revealed several anti-infection drugs, which were also expected because the training set presents several genes/proteins associated with immune response. Hence, the chart may be useful for inferring drug classes or drug compounds that could be repurposed for the treatment of NASH.
Our second strategy was based on using the four genes largely and reproducibly associated with NAFLD and NASH (PNPLA3, TM6SF2, GCKR, and HSD17B13) to predict curated chemicalŌĆōgene/protein interactions in the Comparative Toxicogenomics Database available at (http://ctdbase.org) (Fig. 7B). The aim of this approach was to infer and/or uncover potential associated disease mechanisms from genetic predisposing factors that can yield biologically informative insights. Notably, some drugs were consistently found to interact with three of the loci, for example valproic acid (valproate) (Fig. 7B).
Valproate appears to impact on fatty acid metabolism, and the use of valproate has been linked to the development of obesity and probably NAFLD [44]. Furthermore, valproate acts as a direct histone deactylase (HDAC) inhibitor [45]. While tissue-specific DNA methylation in NAFLD and NASH, including 5-hydroxymethylcytosine (5-hmC) has been previously studied [46-48], the role of other epigenetic mechanisms, including acetylation and deacetylation of histones, remains to be fully ascertained [2]. Likewise, the use of valproate has been associated with the inhibition of mitochondrial beta-oxidation and peroxisomal stimulation in rodent livers [49], which reinforces the concept that the progression of NAFLD into severe clinical and histological forms involves mitochondrial dysfunction [48,50-52]. Table 1 provides a complete list of predicted valproic acid-KEGG pathways, which involve alanine, aspartate and glutamate metabolism, arachidonic acid metabolism, retinol metabolism, PPAR and insulin signaling pathway, regulation of actin cytoskeleton, and hedgehog signaling pathway, among many other pathways relevant to the NAFLD pathogenesis [12]. In addition, the list of valproate-linked pathways contains propanoate and butanoate metabolism that has been linked to the NASH-associated tissue microbiome [12].
Another compound that appears to be linked to three of the genes (PNPLA3, TM6SF2, and GCKR) is quercetin, an antioxidant phenolic heterocyclic compound that is a specific quinone reductase 2 (QR2) inhibitor (Fig. 7B). Collectively, these genetic-chemical interactions provide valuable information about cellular and biological mechanisms of disease, suggesting that this strategy may be used as a complement in the target-based drug development as well.
LIMITATIONS OF THE SYSTEMS BIOLOGY APPROACH
Some limitations of the systems biology approach must be highlighted, including the restricted possibility of adequately addressing the gender dimension of the disease. Sexual dimorphism is observed not only in the prevalence of NAFLD but also in the disease pathogenesis and diverse histological outcomes [53,54]. Although sex differences substantially contribute to the biology of NAFLD, there is limited information on the sex-specific genetic architecture of the disease. We have addressed this aspect by meta-regression analysis of studies that assessed the effect of rs738409 on NAFLD, and we found a negative correlation between the male proportion in the studied populations and the effect of the SNP on liver fat content [19], suggesting that sexual dimorphism might be involved in the impact of the variant on NAFLD development. Likewise, a recent study that involved large-scale analysis of transcriptomic profiles from human livers suggested the sexually dimorphic nature of NASH and its link with fibrosis and responses to drugs [55].
Unrevealing the genetic mechanisms that contribute to sex-specific NAFLD risk should be urgently addressed in further candidate gene association or GWAS studies of NAFLD. Elucidating the sexspecific genetic architecture of NAFLD represents an important area for future research. Besides, there are many other important genetic and epigenetic factors [2,46,48], including mitochondrial genetics [51,56], that play a substantial role in the disease biology, and that deserve a more detailed analysis.
CONCLUSIONS
The utility of the systems biology approach for accelerating the NASH drug discovery process remains to be established. Still, to attain its full potential, refined genomic strategies must be implemented to increase knowledge of genetic susceptibility across all ancestry groups. Although theoretically powerful, PRSs need to be validated and built, not only on validated GWAS-variants but also on robust and global genetic information, preferably specific to each ethnic group. Finally, disciplines such as chemical genetics must be used in tandem with traditional drug discovery approaches to accelerate the progress of precision medicine in NASH and to reveal the druggable NASH genome/proteome.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Supplement
Supplement Print
Print





