

INTRODUCTION
In mammals, liver is a central organ for controlling the metabolic homeostasis for carbohydrates, lipid, and proteins. Dysregulation of liver functions could lead to the metabolic disorder that is ultimately detrimental to the health of individuals. Recently, the increased incidence of obesity is recognized as a major cause for the promotion of metabolic diseases including non-alcoholic fatty liver diseases (NAFLD), which is not only linked with other metabolic diseases such as diabetes, but also invoke more severe liver diseases including non-alcoholic steatohepatitis (NASH), hepatic cirrhosis, and hepatocellular carcinoma (HCC).1
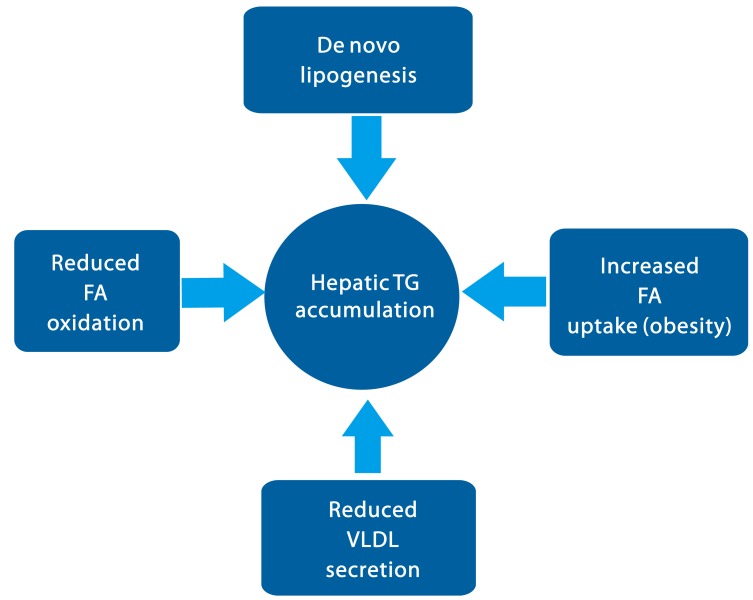
NAFLD can be characterized by the increased accumulation of lipid in the liver, which can be stemmed from the multiple factors. Increased lipolysis from the fat cells or the increased intake of dietary fat, followed by the enhancement of free fatty acids (FFA) can explain this phenomenon.2 Mitochondrial dysfunction that is associated with insulin resistance, which normally precedes the NAFLD, could also cause lipid accumulation by impairment of fatty acid beta oxidation.3 In addition, de novo lipogenesis in the liver contributes greatly to the hepatic steatosis.4 Finally, reduction in lipid clearance that is often associated with insulin resistance can also exacerbate the condition (Fig. 1).5
Accumulation of lipid in the liver can further stimulate existing hepatic insulin resistance by generation of lipid-derived second messengers such as diacylglycerol (DAG) and ceramides.6 Furthermore, lipid accumulation in the liver is also linked with the progression of endoplasmic reticulum stress (ER stress), mitochondria stress, and impaired autophagy, resulting in the condition known as lipotoxicity.7 This latter event can cause the immune response in the Kupffer cells and hepatic stellate cells, which leads to the progression of NASH, hepatic cirrhosis, and in some severe cases, hepatocellular carcinoma.
In this review, we would like to delineate the molecular mechanism for lipid accumulation in the liver as a major precursor for the NAFLD. In particular, we will delineate the individual mechanisms for the triglycerides (TG) synthesis and clearance that is critical in mediating lipid homeostasis in the liver both under physiological conditions and pathological conditions. Understanding of the molecular basis of these pathways could shed the insight into the potential therapeutics in the treatment of this disease.
Fatty acids uptake
Free fatty acids (FFA) in the plasma can be taken up by the liver, and serve as important sources for the TG synthesis in the liver. Normally, plasma FFA is generated by white adipocytes via lipolysis, which is induced by beta adrenergic receptor agonists such as catecholamine under fasting conditions.8 This process involves the regulation of protein kinase A (PKA)-dependent phosphorylation and activation of hormone-sensitive lipase (HSL), a key rate limiting enzyme in the lipolysis, to promote this pathway. This pathway is reversed by insulin under feeding conditions, limiting the liberation of FFA and rather inducing de novo lipogenesis in this tissue. Upon insulin resistance that is associated with obesity, lipolysis is hyperactivated in adipocytes, resulting in the increases in plasma FFA.9 In addition, since obesity is often associated with increased uptake of nutrition rich in lipid, we would expect to observe higher levels of precursors for TG synthesis in the liver.
The main plasma membrane transporters for FFA are fatty acid transporter protein (FATP), caveolins, fatty acid translocase (FAT)/CD36, and fatty acid binding protein (FABP). In mammals 6 members of FATPs are found that contain a common motif for fatty acid uptake and fatty acyl-CoA synthetase function.10 Among family members, FATP2 and FATP5 are highly expressed in the liver, and utilized as major FATPs for the normal physiological context. Indeed, FATP5 knockout mice showed resistance to diet-induced obesity and hepatic TG accumulation, although no clinical evidence for the involvement of this FATP isoform in human obesity.11 Caveolins consist of three protein family members termed caveolins 1, 2, and 3, and are found in the membrane structures called caveolae, which are important for protein trafficking and the formation of lipid droplets.12 Caveolin-1 knockout mice exhibited lower TG accumulation in the liver and showed resistance to diet-induced obesity, showing the importance of this protein in the TG synthesis under obesity. FAT/CD36 is a transmembrane protein that accelerates FA uptake via facilitated diffusion.13 Normally, this protein is not highly expressed in the liver, but is enhanced by diet-induced obesity. The hepatic expression of CD36 was positively correlated with hepatic TG contents in NAFLD patients, underscoring the potential importance of this transporter with this disease.14 FABPs are cytosolic lipid binding proteins that facilitate intracellular transport of FFAs.15 Among 9 isoforms, FABP1 and FABP5 are highly expressed in the liver. Mice with liver-specific deletion of FABP1 displayed resistance to diet-induced obesity, although hepatic TG accumulation did not differ from that of wild type mice, suggesting that contributions from other FABPs might be critical in hepatic TG accumulation in this state.16 Indeed, expression of FABP4 and FABP5 in the liver was correlated with hepatic fatty infiltration in NAFLD patients.17 Further study is necessary to integrate roles of these fatty acid transporters in the hepatic FFA uptake under the physiological conditions and the pathological conditions.
De novo lipogenesis
De novo lipogenesis is an integrated metabolic pathway that comprises of glycolysis (conversion of glucose to acetyl-CoA), biosynthesis of saturated fatty acid followed by desaturation, and the formation of TG. Key rate limiting enzymes in the process include glucokinase and liver-type pyruvate kinase in the glycolysis, acetyl-CoA carboxylase (ACC) and fatty acid synthase (FAS) in the fatty acid synthesis, long chain fatty acid elongase 6 (ELOVL6) and stearoyl-CoA desaturase (SCD) in the formation of monounsaturated fatty acids, and glycerol-3-phosphate acyltransferase (GPAT), lipins, and acyl-CoA: diacylglycerol acyltransferase (DGAT) in the formation of TG.18
FAS is a rate-limiting enzyme in the fatty acid biosynthesis and catalyzes the last step in this pathway.19 Liver-specific knockout of FAS in mice, however, displayed fatty liver phenotypes upon high carbohydrate diet, perhaps due to the increase in hepatic malonyl-CoA contents that would inhibit fatty acid beta oxidation.20 ACC not only catalyzes a key rate-limiting step in the fatty acid biosynthesis, but also is involved in the control of fatty acid oxidation by synthesis of malonyl-CoA, an inhibitor for carnitine palmitoyl transferase 1 (CPT1).21 Indeed, inhibition of liver-specific isoform ACC1 in mice reduced hepatic TG levels in mice, by simultaneously inhibiting fatty acid biosynthesis and activating fatty acid beta oxidation in the liver.19 SCD1 is a microsomal enzyme that catalyses the formation of monounsaturated long-chain fatty acids from saturated fatty acyl-CoAs (E.g. conversion of palmitoyl-CoA (16:0) to palmitoleoyl-CoA (16:1 n-7), and stearoyl-CoA (18:0) to oleoyl-CoA (18:1 n-9)), and is predominantly expressed in the liver.22 Depletion of SCD1 in mice resulted in the reversal of hepatic steatosis under western diet due to the decreased lipogenesis and increased fatty acid beta oxidation.23 DGAT catalyzes the final step of TG synthesis by catalyzing the acylation of diacylglycerol (DAG).24 Inactivation of hepatic isoform DGAT2 in obese mice resulted in the significant reduction in hepatic TG contents, showing the in vivo evidence for the importance of this protein in the de novo lipogenesis.25
Two major transcription factors for lipogenesis, sterol regulatory element binding protein 1c (SREBP-1c) and carbohydrate response element binding protein (ChREBP), are involved in the transcriptional activation of genes encoding aforementioned rate-limiting enzymes in the lipogenesis, and have been associated with increased de novo lipogenesis in NAFLD.4 SREBP-1c is a member of SREBP family that control transcriptional regulation of lipid metabolism.26 As ER-bound precursors, full-length SREBPs reside in the ER by using its transmembrane domain in the middle. Transport of SREBPs from the ER to the Golgi apparatus is mediated in part by nutrient sensors SCAP and Insig, and the mature form of SREBPs is generated by two consecutive proteolytic cleavages. The expression of SREBP-1c is highly induced by insulin under feeding conditions and by saturated fatty acids upon high fat diet feeding in mice, and the liver X receptor alpha (LXR alpha) is shown to be critical in the transcriptional activation of SREBP-1c in this process.27 The activity of SREBP-1c can be further activated by mammalian target of rapamycin (mTOR) pathway, while it can be inhibited by PKA, AMP-activated protein kinase (AMPK), and salt inducible kinases (SIKs).28-30 ChREBP was first identified as a regulator for the hepatic glycolysis by activating transcription of L-PK gene, and was later shown to be involved in the regulation of other lipogenic genes in the pathway.31 At low glucose conditions, ChREBP is present in the cytosol by PKA-mediated phosphorylation, but undergoes dephosphorylation, association with its heterodimeric partner Mlx, and nuclear localization at high glucose conditions, resulting in the transcriptional activation of target genes in the liver. The additional role of AMPK in the regulation of ChREBP was also suggested, although the exact mechanism is still verified in vivo yet. In obese, insulin-resistant ob/ob mice, both SREBP-1c and ChREBP are highly abundant in the liver, and reduction of either factor was shown to be beneficial in relieving hepatic steatosis in mice, underscoring the importance of these lipogenic transcription factors in de novo lipogenesis and the TG accumulation in the liver.32,33
Fatty acid oxidative pathways
Fatty acid beta oxidation in mitochondria is a process to shorten the fatty acids into acetyl-CoA, which can be later converted into ketone bodies (beta hydroxybutyrate or acetoacetate) or can be incorporated into the TCA cycle for the full oxidation.34 To initiate the process, fatty acyl-CoAs should be transported across the mitochondrial membranes by activity of a couple of CPTs. Fatty acyl-CoAs are converted to fatty acyl-carnitines by CPT1 in the mitochondrial outer membrane for the translocation into the intermembrane space. Fatty acyl-carnitines are then transported across the mitochondrial outer membrane by carnitine acylcarnitine translocase. CPT2 converts fatty acyl-carnitines to fatty acyl-CoAs for the fatty acid beta oxidation inside the mitochondrial matrix. The first step involves the beta dehydrogenation of the acyl-CoA ester by chain length-specific acyl-CoA dehydrogenases (e.g. VLCAD, LCAD, and MCAD). Indeed, mice deficient in MCAD and VLCAD develop hepatic steatosis, supporting the role of these proteins and the fatty acid beta oxidation in the hepatic TG content.35 2-enoyl-CoA hydratase, 3-hydroxyacyl-CoA dehydrogenase and 3-oxoacyl-CoA thiolase are subsequently involved in the fatty acid beta oxidation process to complete the conversion of acyl-CoA ester into acetyl-CoA.
Under fasting conditions, fatty acid beta oxidation is enhanced in part via inactivation of ACC, resulting in the reduced production of malonyl-CoA that serves as a potent inhibitor of CPT1.21 Chronic starvation also increases expression of beta oxidation genes via transcriptional mechanisms. Peroxisome proliferator-activated receptor (PPAR) alpha and its co-activator PPAR gamma co-activator 1 (PGC-1) alpha are critical in enhancing expression of target genes including CPT1, LCAD, MCAD, and acyl-CoA oxidase (ACOX).36 Starvation-dependent activation of AMPK and sirtuins could also enhance expression of these genes by directly modifying and activating PGC-1 alpha.37,38 The clinical implication of impaired mitochondrial beta oxidation on the progression of NAFLD is not conclusive, and contradicting reports have been published. Further study is necessary to delineate the role of fatty acid beta oxidation on hepatic lipid accumulation and the progression of NAFLD.39
TG secretion
In the liver, TG secretion is achieved via the formation of very low density lipoprotein (VLDL).40 VLDL consists of hydrophobic core lipids containing TGs and cholesterol esters, which is covered by hydrophilic phospholipids and apolipoprotein B (apoB) 100. ApoB 100 is a liver-specific ApoB that is critical in the VLDL assembly, while apoB 48 in the intestine is associated with chylomicron formation. The VLDL assembly process occurs initially in the rough ER during the translation and translocation of the apoB 100, resulting in the formation of a partially lipidated apoB 100. The partially lipidated apoB 100, termed the pre-VLDL is then transported into the Golgi for the maturation, and subsequently released from the liver via exocytosis. Indeed, hepatic steatosis was reported in patients carrying mutations in apoB 100 (hypobetalipoproteinemia) and in MTP (abetalipoproteinemia), underscoring the importance of these proteins in the lipid homeostasis in humans.41,42
Anabolic hormone insulin is critical in the regulation of VLDL assembly and secretion. Insulin plays a role in the degradation of apoB 100, perhaps utilizing an autophagy-dependent pathway.43 Furthermore, insulin inhibits the transcription of MTP, by phosphatidylinositol 3 kinase/Akt-dependent regulation. Akt phosphorylates and inhibits transcription factor FoxA2, a critical forkhead box factor for activating expression of MTP at the transcription level.44,45 Upon insulin resistance, perturbation of this process results in hypertriglyceridemia via increased TG secretion. However, the rate of TG secretion cannot keep up with the increased rate of TG synthesis in this condition, resulting in the hepatic steatosis in spite of the increased VLDL secretion. Similar phenotype is observed in NAFLD patients, which exhibit both hypertriglyceridemia and hepatic steatosis.46 Prolonged exposure of the liver to FFA would promotes ER stress and other oxidative stress in the liver, leading to the degradation of apoB 100, decrease in the VLDL secretion, and worsening of hepatic steatosis.
CONCLUSIONS
The balance between the TG uptake/synthesis and TG hydrolysis/secretion is critical in the maintenance of lipid homeostasis in the liver. Clearly, perturbation of any of these pathways could be detrimental to the lipid metabolism. In the case of NAFLD, the progression of hepatic steatosis can stem from the increased FFA uptake and de novo lipogenesis for the increased TG synthesis, and the decreased TG hydrolysis and fatty acid beta oxidation. Reduced TG secretion via VLDL could also promote hepatic lipid accumulation, although the VLDL secretion was rather increased in NAFLD patients. Understanding of molecular mechanisms of each pathway is critical in pursuing the development of therapeutics of NAFLD in the future.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print




