


INTRODUCTION
Alcohol is classified as a group 1 carcinogen by the International Agency for Research on Cancer because it induces hepatocellular carcinoma like as other cancers in humans.
Worldwide, liver cancer is the second highest cause of cancerrelated death in men and the sixth highest cause of cancer-related death in women. Liver cancer is more common in low-income and middle-income countries than in developed countries [1]. According to the National Cancer Institute, approximately 40,700 cases of liver cancer are expected to be newly diagnosed, and approximately 29,000 patients will die from liver cancer in the USA in 2017. Besides, the incidence of liver cancer is increasing by approximately 3–4% per year [2]. Indeed, liver cancer is a major public health problem. Hepatocellular carcinoma (HCC), which accounts for around 70–90% of cases, is the most common type of primary liver cancer. Alcohol consumption, the level of which is higher in developed countries, especially in the USA and Europe, is one of the frequent causes of HCC in developed countries [3]. The ratio of alcohol abuse to all etiologies of HCC varies according to the country and area; alcohol abuse is reported to be responsible for approximately 15–30% of HCC [4] Understanding the clinical features and the mechanisms of alcohol-based HCC is critically important to the prevention and detection of early-stage HCC and for the development of treatments for HCC. This review summarizes the recent clinical and pathological studies investigating the hepatocarcinogenesis in alcohol-related liver disease (ALD).
THE RISKS OF LIVER CIRRHOSIS AND HCC
According to a World Health Organization report, approximately 280 million individuals, or 4.1% of the population aged >15 years, meet the definition of ‘alcohol-use disorder’ (alcohol dependence and the harmful use of alcohol). The prevalence is almost the same as the prevalence of hepatitis B, and is four times higher than the prevalence of hepatitis C [5]. Because of the large population, HCC screening such as ultrasonography or the measurement of serum tumor marker levels for all of such patients would lead to huge medical costs, thus, it is necessary to select individuals with a high risk of HCC. In this respect, the American Association for the Study of Liver Diseases (AASLD) recommends that patients with Child’s classification A/B cirrhosis undergo surveillance for HCC using ultrasonography with or without alpha-fetoprotein measurement, every 6 months, and does not recommend the modification of the surveillance strategy based on the etiology of liver disease, the strategy of which is almost the same as that recommended by the European Association for the Study of the Liver [5]. Incidentally, the AASLD guidelines for the management of HCC suggested that HCC surveillance is cost-effective if the annual incidence of HCC is 1.5% in patients with cirrhosis [6]. Similar to hepatitis C and hepatitis B, the presence of alcoholic liver cirrhosis is considered to be an important risk factor for the development of HCC. It has been reported that approximately 10–20% of heavy drinkers develop cirrhosis [5]. Several previous studies that have assessed the annual incidence of HCC in patients with alcohol-induced liver cirrhosis have revealed the rate to be 1.9–2.6%, thus, it might be appropriate to perform HCC surveillance for patients with alcoholic liver cirrhosis [7]. However, even when guideline-based surveillance was performed, almost 20–30% of HCC in patients with cirrhosis were diagnosed at a non-early stage [8].
PATHOGENESIS AND GENETICS OF ALCOHOLINDUCED LIVER CARCINOGENESIS
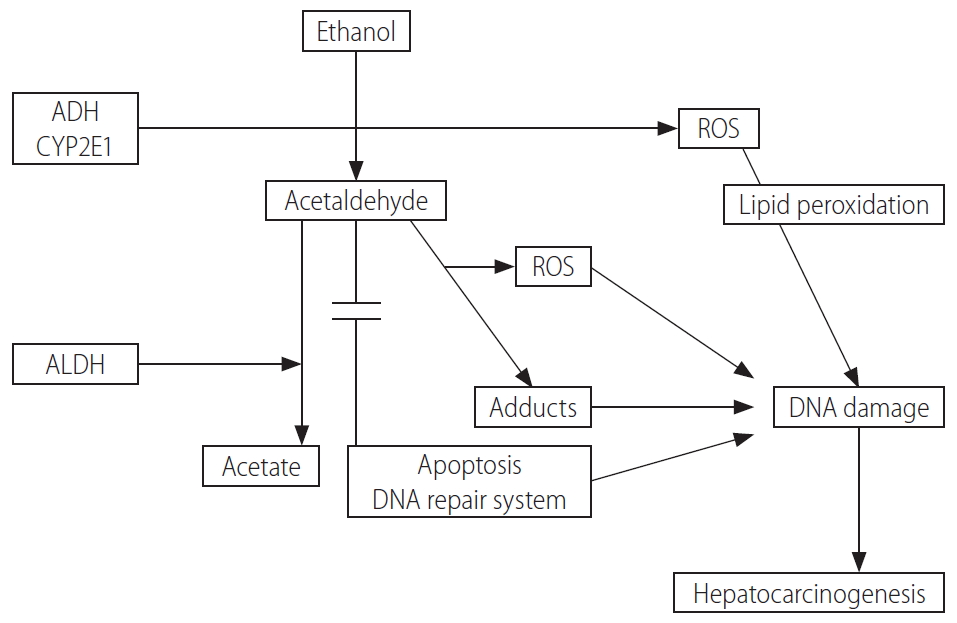
Chronic alcohol intake alters the architecture and compromises the functional capacity of the liver by triggering steatosis, steatohepatitis and cirrhosis [9]. These pathological events are subsequently sustained and participate in the carcinogenetic process. A number of pathophysiological factors are specific to hepatic alcohol-mediated carcinogenesis, including: 1) the formation of acetaldehyde and its direct detrimental effects on proteins and DNA; 2) an elevated production of cytochrome P450 family 2 subfamily E member 1 (CYP2E1)- and/or iron-induced reactive oxygen species (ROS), further aggravated by the impairment of antioxidant defenses and DNA repair mechanisms; 3) changes to the immune system and the induction of chronic inflammation; and 4) interference with methyl group transfer and alterations to gene expression (Fig. 1) [3].
These pathways may be further affected by host-related genetic heterogeneity that could partly explain the difference for inter-individual susceptibility to the development of HCC in patients with alcoholic cirrhosis [3].
HEPATOTOXICITY OF ETHANOL AND TUMOR PROMOTION
Ethanol is oxidized into acetaldehyde by alcohol dehydrogenase (ADH) in the cytosol [10]. Acetaldehyde then enters the mitochondria where it is oxidized to acetate by mitochondrial aldehyde dehydrogenase (ALDH). Acetaldehyde is a highly reactive and directly mutagenic compound which forms various protein and DNA adducts that promote DNA repair failure, lipid peroxidation and mitochondrial damage, and ultimately favor carcinogenesis. Another major pathway of ethanol metabolism includes its oxidation in microsomes by the enzyme CYP2E1, a step that requires nicotinamide adenine dinucleotide phosphate rather than nicotinamide adenine dinucleotide, as for ADH. In parallel, ROS are formed through the metabolism of alcohol by CYP2E1 and the re-oxidation of NADH in the mitochondria [11].
Genetic variations in the alcohol-metabolizing enzymes that modulate the resulting amounts of carcinogenic acetaldehyde have been largely explored as potential inherited markers of alcohol-induced cancers, including those of the liver [12]. In particular, patients with ALD who are homozygous for the ADH1C*1 allele (thought to confer higher enzymatic activity) appear to be more vulnerable to the onset of HCC [13]. Similarly, the weakly active ALDH2*2-conferring allele has been found to be associated with liver cancer in excessive drinkers [14].
ALCOHOL-INDUCED OXIDATIVE STRESS AND IRON METABOLISM
A pivotal mechanism implicated in alcohol-related hepatocarcinogenesis is oxidative stress, which is secondary to ROS derived from alcohol metabolism, inflammation and increased iron storage. Indeed, ROS promote damage to cellular macromolecules and participate in the progression of liver carcinogenesis through the formation of lipid peroxides such as 4-hydroxy-nonenal [15]. An accumulation of ROS causes structural and functional alterations to DNA and affects gene functions such as replication and transcription, and plays a major role in cancer initiation and promotion [16]. ROS accumulation also induces the production of various cytokines, the activation of immune cells and the upregulation of angiogenesis and the metastatic process [17].
ROS include hydroxyethyl, superoxide anion, hydroxyl radicals and numerous free radicals that accumulate following the successive actions of pro- and antioxidant enzymes, while antioxidant defenses are impaired by ethanol consumption, genetic variants affecting the enzymes that regulate the production and detoxification of ROS have been shown to modulate the outcome of ALD. In particular, myeloperoxidase (MPO), which catalyzes the reaction between H2O2 and Cl- to form the highly reactive hypochlorous acid (HOCl) and anion, 37 is affected by a G to A base exchange at position -463 involving its promoter [18]. Additionally, manganese superoxide dismutase (SOD) 2 generates H2O2 within the mitochondria, leading to the formation of highly reactive HOCl [18]. A genetic dimorphism substitutes either alanine (Ala) or valine in the mitochondrial targeting sequence of SOD2 and results in higher mitochondrial activity for the Ala-SOD2 variant. In large prospective cohorts of patients with alcoholic cirrhosis, it was shown that the carriage of 2 G-MPO alleles and/or the possession of at least one Ala-SOD2 allele alone were independent risk factors for the onset of HCC [12]. These variants were also associated with liver iron overload, possibly through enhanced mitochondrial hydrogen peroxide production, a condition which has also been reported to be associated with a risk of HCC in these patients, along with the usual HFE gene mutations [19].
ACTIVATION OF INNATE IMMUNITY, CYTOKINE AND CHEMOKINE SYSTEMS
Alcohol interacts with the immune system and affects tumor immune surveillance, both mechanisms which may relate with tumor development and progression. The innate immune response aims to identify cancerous clones in order to inactivate transforming cells. This response is promoted by inflammatory mediators (chemokines and cytokines) which are produced by various immune cells [18]. Alcohol consumption increases gut permeability and the translocation of bacteria-derived lipopolysaccharide (LPS) from the gut to the liver, in Kupffer cells, LPS interacts with toll-like receptor (TLR) 4, which leads to the production of pro-inflammatory cytokines such as interleukin (IL)-6 and tumor necrosis factor (TNF)-α [18]. These molecules are major pathways involved in hepatocarcinogenesis [20]. Although the precise mechanisms by which pro-inflammatory cytokines promote liver cancer development are not fully understood, their signals regulate gene expression through the signal transducer and activator of transcription 3(STAT3) and nuclear factor-κB (NF-κB) transcription factors. NF-κB, one of the main transcriptional regulators of the inflammatory response, is activated during ALD and increases the production of various pro-inflammatory mediators which include TNF-α, IL-1, IL-6, EGF and TLRs, the latter promote ROS accumulation and activate STAT3, thus participating in cancer development [21]. In patients with ALD, the highly productive cytokine IL-6 -174G allele is associated with HCC, and epidermal growth factor (EGF) promotes cancer growth and invasiveness, and which is subject to functional polymorphism involving an A to G exchange in the 50 untranslated region of the EGF gene [18]. The G allele, resulting in higher transcription levels, has been associated with the presence of HCC in Caucasian populations affected by alcohol- or hepatitis C virus (HCV)-related liver diseases, a finding that was further confirmed by a recent meta-analysis [22].
MODULATION OF LIPID METABOLISM
Alcohol abuse is characterized by an accumulation of fat (mainly triglycerides, phospholipids and cholesterol esters) in hepatocytes. Initially revealed by genome-wide association studies, some single nucleotide polymorphisms (SNPs) might associated with hepatocarcinogenesis (Table 1). A SNP (rs738409 C>G for I148M) in the adiponutrin/patatin-like phospholipase domain containing protein 3 (PNPLA3) protein sequence has rapidly become a well-established genetic factor associated with steatosis and fibrosis in patients with ALD [12]. This genetic variation is considered a loss-of-function mutation that promotes intracellular triglyceride accumulation and lipotoxicity in hepatocytes [18]. The rs738409 (G) allele has been highlighted as a major genetic driver of liver cancer development in patients with alcoholic cirrhosis by numerous European research groups in the context of both large case- control and prospective studies, as well as subsequent meta-analyses [18]. Other polymorphisms affecting genes involved in lipid metabolism have also been proposed [18].
GUT-LIVER AXIS AND HEPATOCARCINOGENESIS
Consolidating evidence arose in recent years indicating that alcohol not only alters the quantitative and qualitative composition of the microbiome, but also induces alterations of the epithelial intestinal barrier with consequent release of bacteria and bacterial products that fuel the inflammatory response in the liver. It has been demonstrated that, upon alcohol exposure, LPS and other bacterial products are released into the circulation and can bind to members of the TLRs family on the cellular membrane of hepatic resident macrophages (Kupffer cells), thereby triggering the production of pro-inflammatory mediators in these cells [23]. Fecal microbiota transplantation and fecal microbiota manipulation via use of prebiotics might represent a valuable therapy against alcoholic liver injury and steatosis [24,25]. Recent pioneering work has identified gut-liver communication and the microbiome as important components involved in the development of ALD [26,27]. The unpublished data from our institute, in alcohol-related cirrhotic patients, status of dysbiosis was associated and with hepatocarcinogenesis and an independent risk factor for developing HCC.
CLINICAL RISK FACTORS
Age is one of the most significant risk factors for many malignancies including HCC in ALD patients [18]. Alcohol consumption has been associated with an increased risk of several malignancies, this risk starting at doses as low as 10 g/1 unit/day [28]. It is an independent risk factor for the development of HCC, with a relative risk of 2.07 for heavy drinkers compared to non-drinkers and the relative risk is also slightly increased in occasional drinkers, while in the setting of non-fibrotic liver F0/F1, heavy alcohol consumption is no longer associated with an HCC risk after adjustment for smoking habits and metabolic syndrome [29].
There might be a gender difference in the volume of alcohol intake that increases the risk of alcohol-induced liver damage and the development of HCC. It has been reported that the risk of developing cirrhosis becomes substantial with the consumption of 60–80 g/day of alcohol for 10 years in men and 20 g/day for 10 years in women [14,23]. In addition, women showed a more rapid progression (20 years) to cirrhosis than men (35 years) [30]. Among individuals who consume more than 80 g/day of alcohol, the risk of HCC development in women has been shown to be almost fivefold higher than that in men [30]. However, the overall prevalence of HCC in women is small compared with that in men. Various mechanisms have been suggested to underlie the higher sensitivity of women to alcohol. After the oral administration of alcohol, women show less first-pass metabolism of alcohol, which is defined as the difference in the amount of orally administered ethanol and the quantities in the systemic blood, due to their lower gastric ADH activity, which results in a higher serum concentration of alcohol [31]. Thus, even when the same amount of ethanol is consumed, the female liver may be exposed to more ethanol. Furthermore, estrogen, a female sex hormone, may play an important role in alcohol-induced liver injury. It has been shown that estrogen increases the sensitivity of Kupffer cells to LPS, which results in more severe liver injury, and many previous studies have reported that more severe inflammatory responses in the liver and fat tissue, which were associated with TLR4 signaling, were seen in female patients [5].
In patients with ALD, the coexistence of hepatitis virus has been shown to accelerate the disease course. In patients with a high alcohol intake (>60 g/day to 125 g/day), the coexistence of HCV has been shown to increase the risk of alcohol-associated liver cirrhosis [5]. Furthermore, heavy alcohol consumption has also been shown to increase the risk of developing HCC [5]. Patients with coexisting hepatitis B virus (HBV) are at increased risk of developing fibrosis and HCC [5]. In addition, self-resolved HBV infection can be a risk factor for developing HCC in patients with alcoholic cirrhosis [5].
Moreover, alcohol synergizes with other risk factors for HCC, such as diabetes mellitus and viral hepatitis. In patients consuming excessive alcohol, defined as over 80 g/day, the risks of HCC rose from 2.4 to 9.9 in patients with diabetes, and from 19.1 to 53.9 in patients with HCV infection [32,33]. Obesity also has a synergistic effect [34].
Conversely, alcohol cessation is associated with a risk of HCC that falls by 6–7% per year, but the detrimental effects of alcohol can remain for decades, a wash-out period of 23 years being necessary to achieve the same incidence of HCC seen in abstinent patients. However, these results need to be considered with caution as this analysis only included four studies [35].
CONCLUSION
Several clinical factors increase the risk of alcohol-induced HCC. A large alcohol intake, coexistence of diabetes and hepatitis virus infection, and female gender are established factors. For the mechanisms of hepatocarcinogenesis are increasing evidence to suggest that many factors are involved. Alcohol metabolites and adducts have been shown to induce oxidative stress, direct mutagenesis, the aberrant methylation of DNA or protein on hepatocytes, and the immune system might be implicated in the development and progression of HCC. Further studies are needed to reduce the risk of HCC.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print





